

The BCL-2 arbiters of apoptosis and their growing role as cancer targets
- PMID: 29099483
- PMCID: PMC5729526
- DOI: 10.1038/cdd.2017.161
The BCL-2 arbiters of apoptosis and their growing role as cancer targets
Abstract
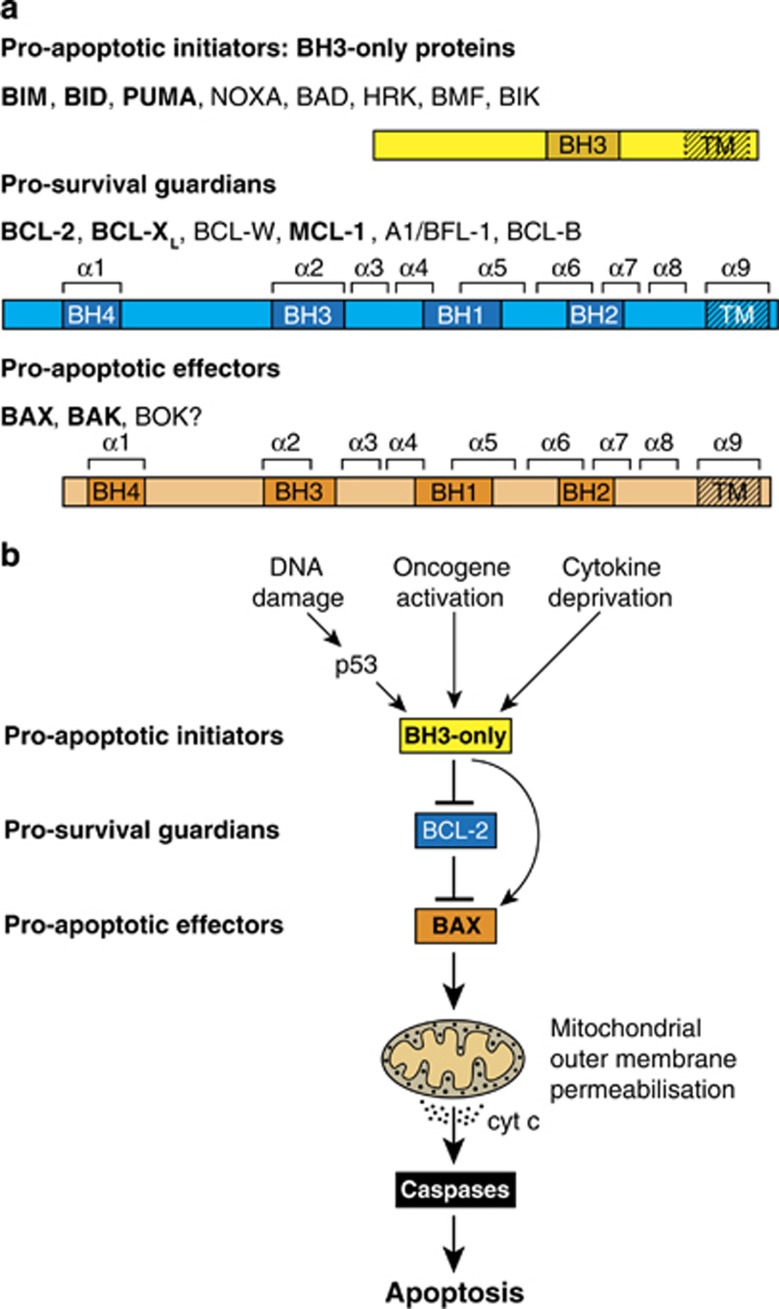
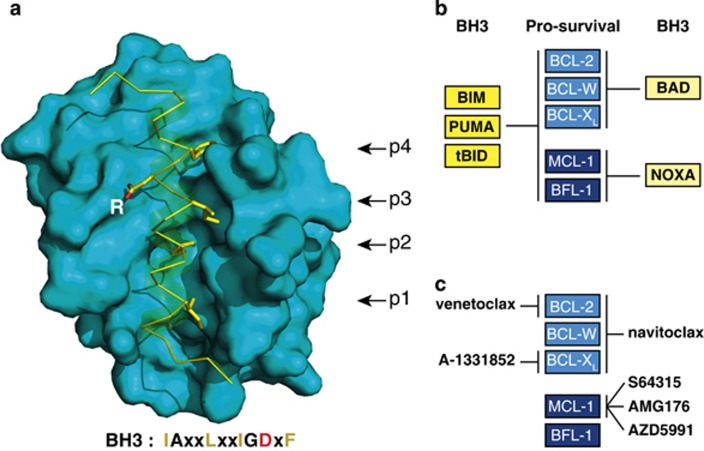
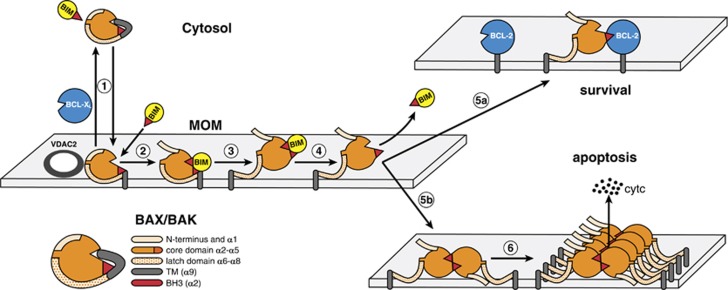
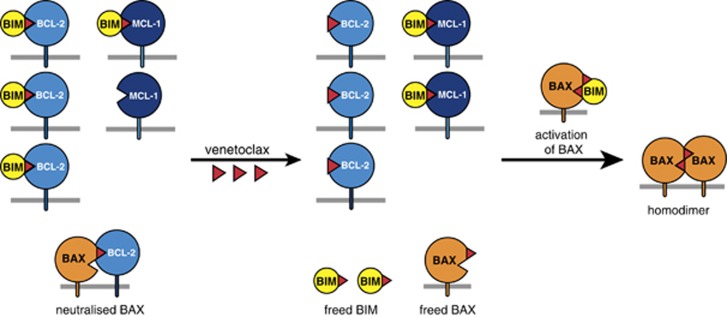
Impaired apoptosis plays a central role in cancer development and limits the efficacy of conventional cytotoxic therapies. Deepening understanding of how opposing factions of the BCL-2 protein family switch on apoptosis and of their structures has driven development of a new class of cancer drugs that targets various pro-survival members by mimicking their natural inhibitors, the BH3-only proteins. These 'BH3 mimetic' drugs seem destined to become powerful new weapons in the arsenal against cancer. Successful clinical trials of venetoclax/ABT-199, a specific inhibitor of BCL-2, have led to its approval for a refractory form of chronic lymphocytic leukaemia and to scores of on-going trials for other malignancies. Furthermore, encouraging preclinical studies of BH3 mimetics that target other BCL-2 pro-survival members, particularly MCL-1, offer promise for cancers resistant to venetoclax. This review sketches the impact of the BCL-2 family on cancer development and therapy, describes how interactions of family members trigger apoptosis and discusses the potential of BH3 mimetic drugs to advance cancer therapy.
Conflict of interest statement
WEHI has received research funding from Genentech and AbbVie, and royalty and milestone payments for venetoclax.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
