

Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes
- PMID: 29100084
- PMCID: PMC5673627
- DOI: 10.1016/j.ajhg.2017.09.013
Profiling of Short-Tandem-Repeat Disease Alleles in 12,632 Human Whole Genomes
Abstract
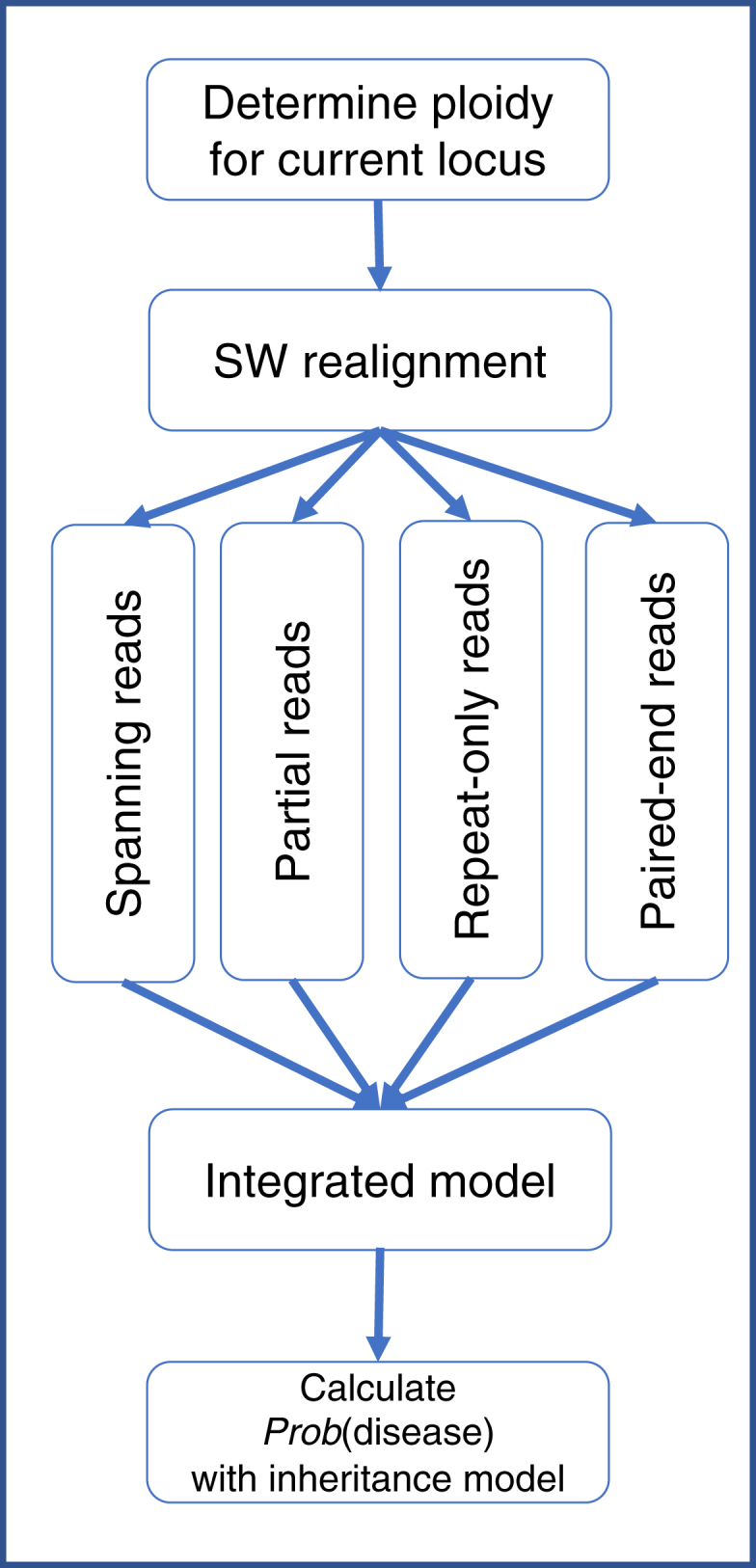
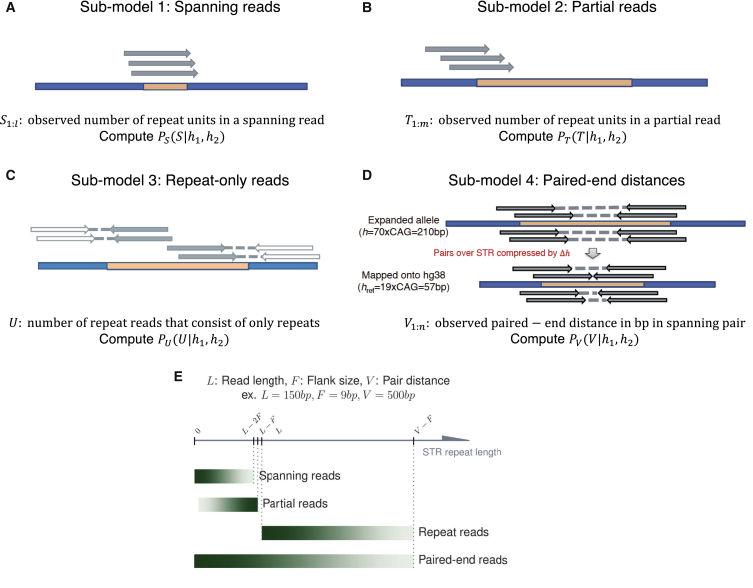
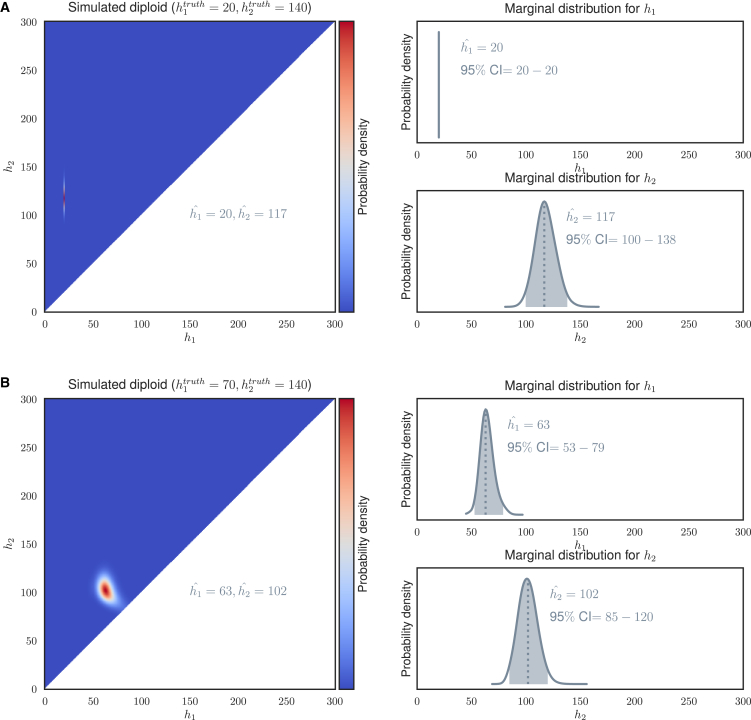
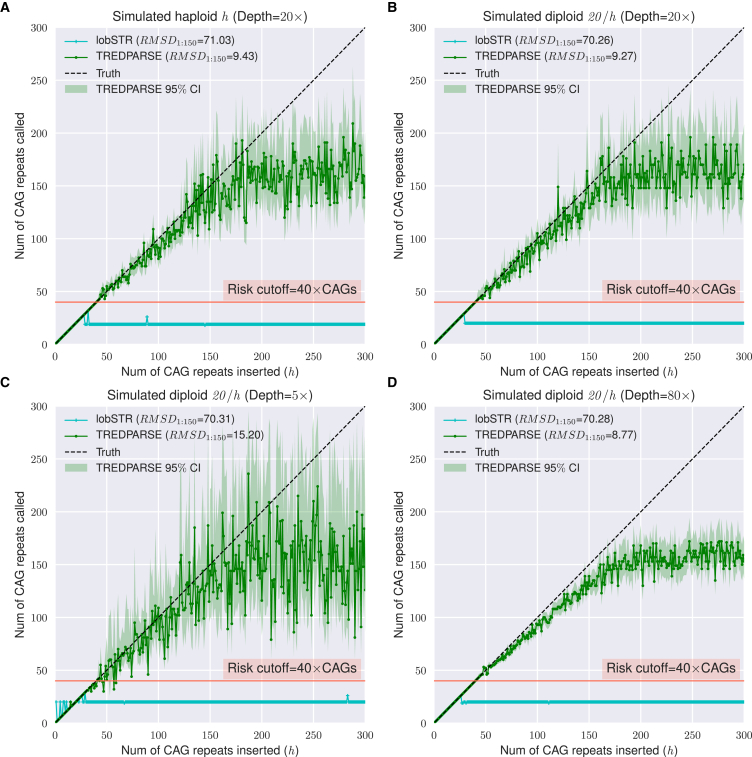
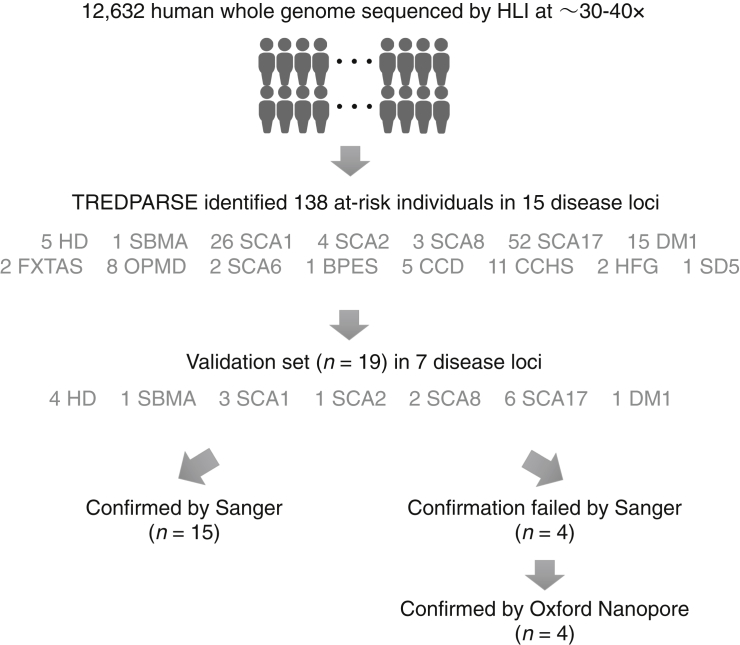
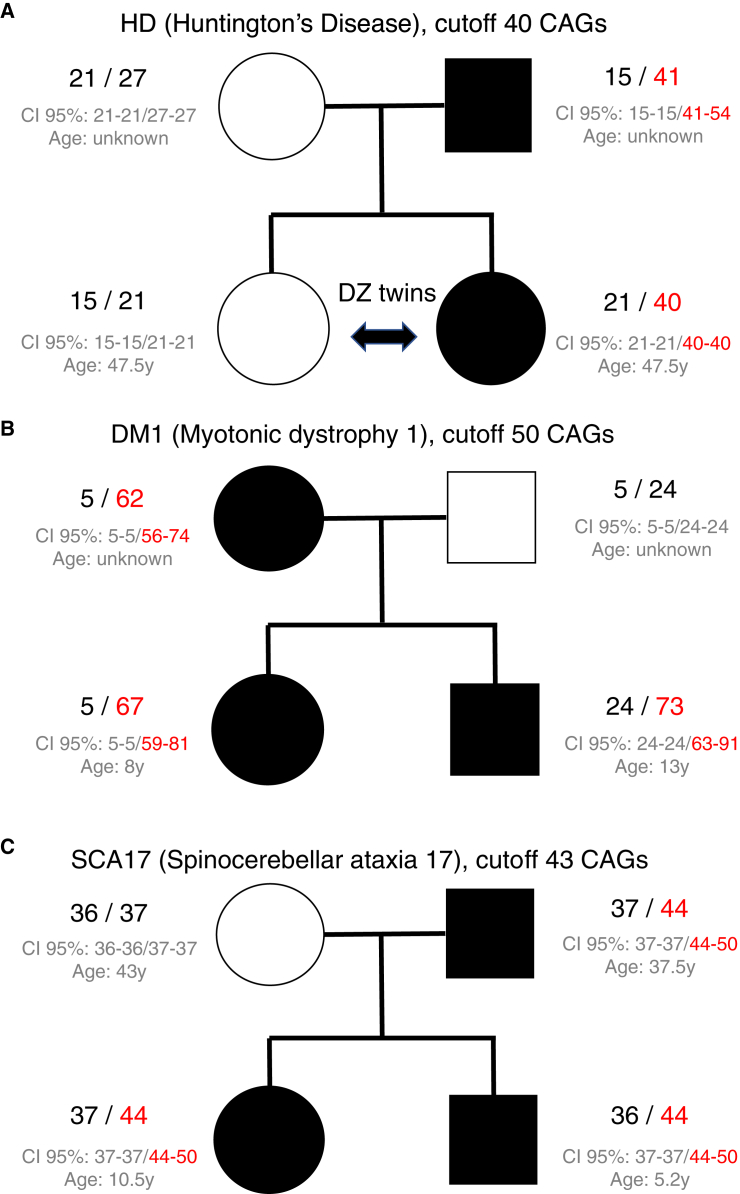
Short tandem repeats (STRs) are hyper-mutable sequences in the human genome. They are often used in forensics and population genetics and are also the underlying cause of many genetic diseases. There are challenges associated with accurately determining the length polymorphism of STR loci in the genome by next-generation sequencing (NGS). In particular, accurate detection of pathological STR expansion is limited by the sequence read length during whole-genome analysis. We developed TREDPARSE, a software package that incorporates various cues from read alignment and paired-end distance distribution, as well as a sequence stutter model, in a probabilistic framework to infer repeat sizes for genetic loci, and we used this software to infer repeat sizes for 30 known disease loci. Using simulated data, we show that TREDPARSE outperforms other available software. We sampled the full genome sequences of 12,632 individuals to an average read depth of approximately 30× to 40× with Illumina HiSeq X. We identified 138 individuals with risk alleles at 15 STR disease loci. We validated a representative subset of the samples (n = 19) by Sanger and by Oxford Nanopore sequencing. Additionally, we validated the STR calls against known allele sizes in a set of GeT-RM reference cell-line materials (n = 6). Several STR loci that are entirely guanine or cytosines (G or C) have insufficient read evidence for inference and therefore could not be assayed precisely by TREDPARSE. TREDPARSE extends the limit of STR size detection beyond the physical sequence read length. This extension is critical because many of the disease risk cutoffs are close to or beyond the short sequence read length of 100 to 150 bases.
Keywords: genetic disorder; genome sequencing; genotyping; microsatellites; population genetics; short tandem repeats; trinucleotide repeat expansion.
Copyright © 2017 The Author(s). Published by Elsevier Inc. All rights reserved.
Figures
References
-
- Zhivotovsky L.A., Underhill P.A., Cinnioğlu C., Kayser M., Morar B., Kivisild T., Scozzari R., Cruciani F., Destro-Bisol G., Spedini G. The effective mutation rate at Y chromosome short tandem repeats, with application to human population-divergence time. Am. J. Hum. Genet. 2004;74:50–61. - PMC - PubMed
-
- Helgason A., Einarsson A.W., Guðmundsdóttir V.B., Sigurðsson Á., Gunnarsdóttir E.D., Jagadeesan A., Ebenesersdóttir S.S., Kong A., Stefánsson K. The Y-chromosome point mutation rate in humans. Nat. Genet. 2015;47:453–457. - PubMed
-
- Hares D.R. Selection and implementation of expanded CODIS core loci in the United States. Forensic Sci. Int. Genet. 2015;17:33–34. - PubMed
-
- Gymrek M., McGuire A.L., Golan D., Halperin E., Erlich Y. Identifying personal genomes by surname inference. Science. 2013;339:321–324. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
