

RECQ1 expression is upregulated in response to DNA damage and in a p53-dependent manner
- PMID: 29100281
- PMCID: PMC5652675
- DOI: 10.18632/oncotarget.18237
RECQ1 expression is upregulated in response to DNA damage and in a p53-dependent manner
Abstract
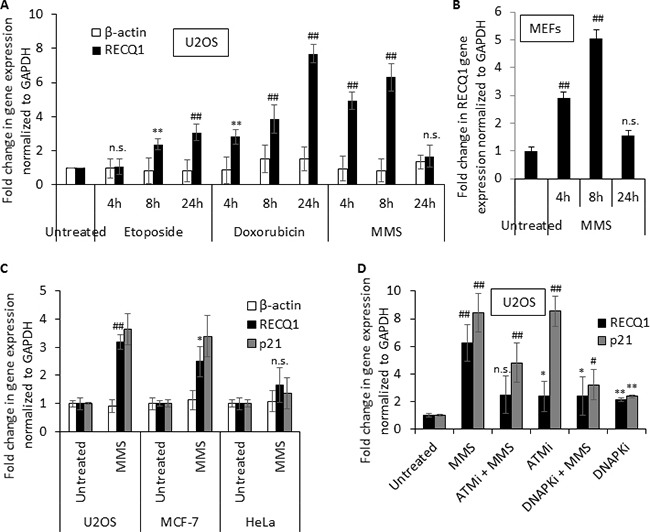
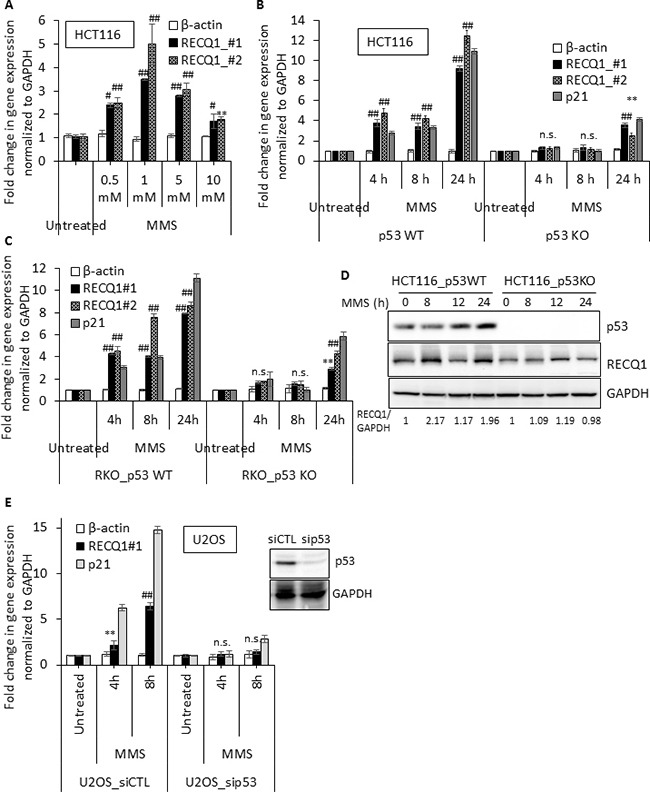
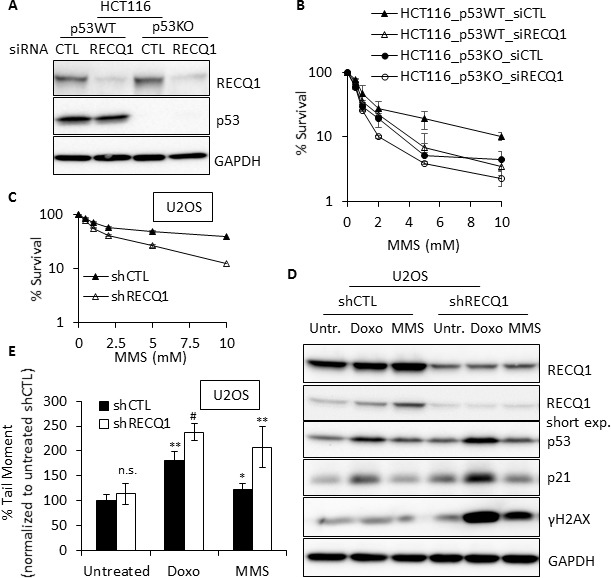
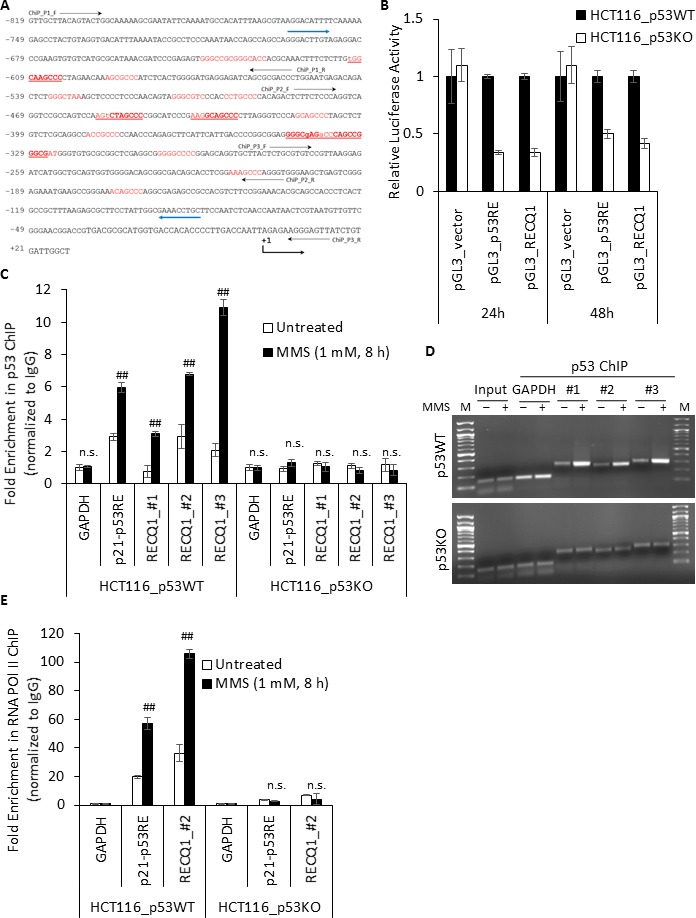
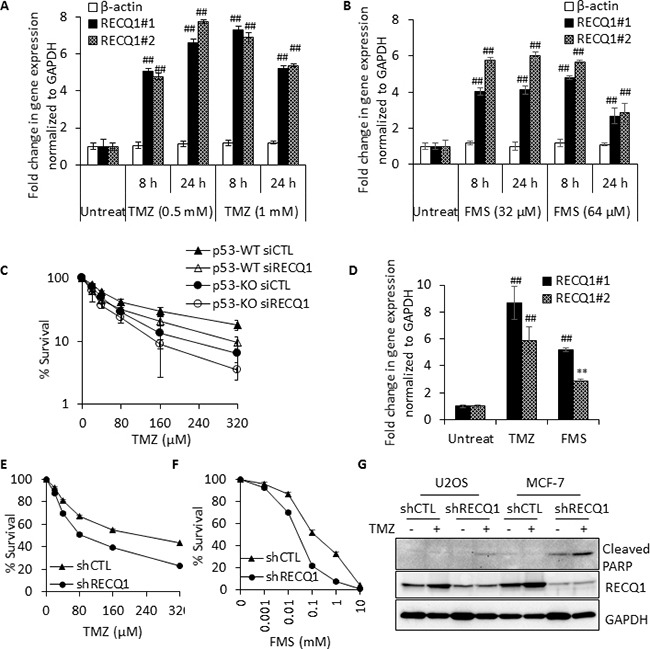
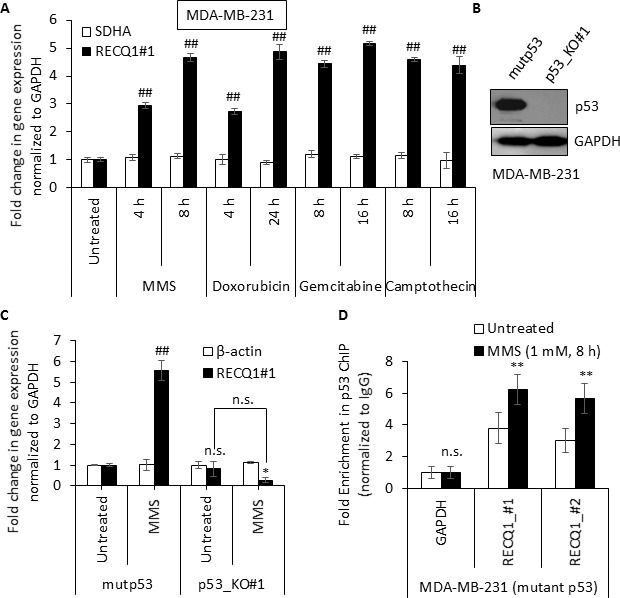
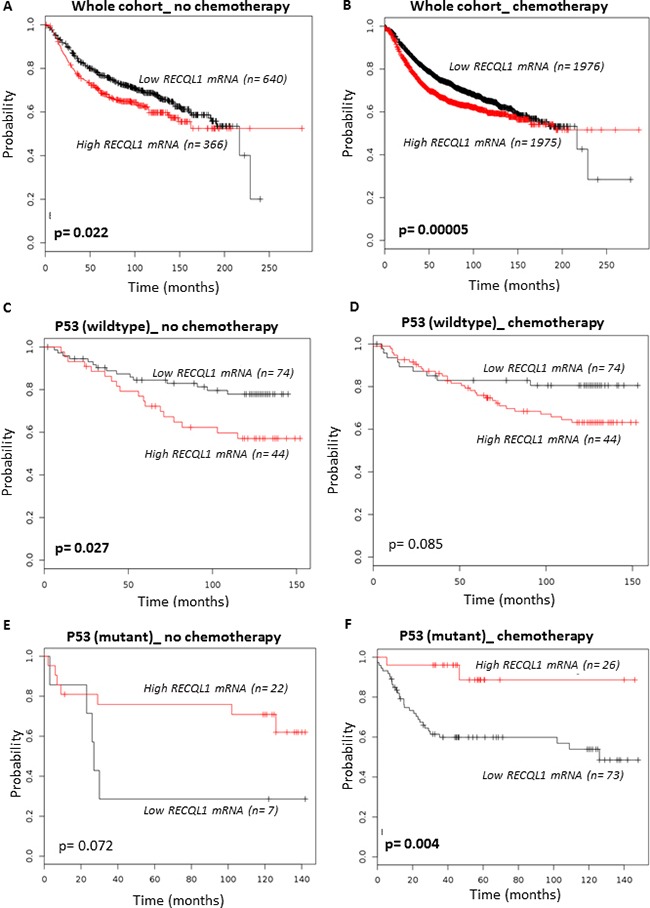
Sensitivity of cancer cells to DNA damaging chemotherapeutics is determined by DNA repair processes. Consequently, cancer cells may upregulate the expression of certain DNA repair genes as a mechanism to promote chemoresistance. Here, we report that RECQ1, a breast cancer susceptibility gene that encodes the most abundant RecQ helicase in humans, is a p53-regulated gene, potentially acting as a defense against DNA damaging agents. We show that RECQ1 mRNA and protein levels are upregulated upon treatment of cancer cells with a variety of DNA damaging agents including the DNA-alkylating agent methylmethanesulfonate (MMS). The MMS-induced upregulation of RECQ1 expression is p53-dependent as it was observed in p53-proficient but not in isogenic p53-deficient cells. The RECQ1 promoter is bound by endogenous p53 and is responsive to p53 in luciferase reporter assays suggesting that RECQ1 is a direct target of p53. Treatment with the chemotherapeutic drugs temozolomide and fotemustine also increased RECQ1 mRNA levels whereas depletion of RECQ1 enhanced cellular sensitivity to these agents. These results identify a previously unrecognized p53-mediated upregulation of RECQ1 expression in response to DNA damage and implicate RECQ1 in the repair of DNA lesions including those induced by alkylating and other chemotherapeutic agents.
Keywords: DNA damage; RecQ; gene expression; helicase; p53.
Conflict of interest statement
CONFLICTS OF INTERESTS The authors do not have any conflicts of interest.
Figures
Similar articles
-
The DNA repair helicase RECQ1 has a checkpoint-dependent role in mediating DNA damage responses induced by gemcitabine.J Biol Chem. 2019 Oct 18;294(42):15330-15345. doi: 10.1074/jbc.RA119.008420. Epub 2019 Aug 23. J Biol Chem. 2019. PMID: 31444271 Free PMC article.
-
Identification of RECQ1-regulated transcriptome uncovers a role of RECQ1 in regulation of cancer cell migration and invasion.Cell Cycle. 2014;13(15):2431-45. doi: 10.4161/cc.29419. Cell Cycle. 2014. PMID: 25483193 Free PMC article.
-
Transcriptome guided identification of novel functions of RECQ1 helicase.Methods. 2016 Oct 1;108:111-7. doi: 10.1016/j.ymeth.2016.04.018. Epub 2016 Apr 18. Methods. 2016. PMID: 27102625 Free PMC article.
-
The potential roles of p53 tumor suppressor in nucleotide excision repair (NER) and base excision repair (BER).Exp Mol Med. 2004 Dec 31;36(6):505-9. doi: 10.1038/emm.2004.64. Exp Mol Med. 2004. PMID: 15665582 Review.
-
MGMT: key node in the battle against genotoxicity, carcinogenicity and apoptosis induced by alkylating agents.DNA Repair (Amst). 2007 Aug 1;6(8):1079-99. doi: 10.1016/j.dnarep.2007.03.008. Epub 2007 May 7. DNA Repair (Amst). 2007. PMID: 17485253 Review.
Cited by
-
Diepoxybutane induces the expression of a novel p53-target gene XCL1 that mediates apoptosis in exposed human lymphoblasts.J Biochem Mol Toxicol. 2020 Mar;34(3):e22446. doi: 10.1002/jbt.22446. Epub 2020 Jan 18. J Biochem Mol Toxicol. 2020. PMID: 31953984 Free PMC article.
-
The DNA repair helicase RECQ1 has a checkpoint-dependent role in mediating DNA damage responses induced by gemcitabine.J Biol Chem. 2019 Oct 18;294(42):15330-15345. doi: 10.1074/jbc.RA119.008420. Epub 2019 Aug 23. J Biol Chem. 2019. PMID: 31444271 Free PMC article.
-
Transcription/Replication Conflicts in Tumorigenesis and Their Potential Role as Novel Therapeutic Targets in Multiple Myeloma.Cancers (Basel). 2021 Jul 27;13(15):3755. doi: 10.3390/cancers13153755. Cancers (Basel). 2021. PMID: 34359660 Free PMC article. Review.
-
RECQ1 Helicase in Genomic Stability and Cancer.Genes (Basel). 2020 Jun 5;11(6):622. doi: 10.3390/genes11060622. Genes (Basel). 2020. PMID: 32517021 Free PMC article. Review.
-
Fanconi anemia-independent DNA inter-strand crosslink repair in eukaryotes.Prog Biophys Mol Biol. 2020 Dec;158:33-46. doi: 10.1016/j.pbiomolbio.2020.08.005. Epub 2020 Aug 30. Prog Biophys Mol Biol. 2020. PMID: 32877700 Free PMC article. Review.
References
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
