

Immunohaematological complications in patients with sickle cell disease after haemopoietic progenitor cell transplantation: a prospective, single-centre, observational study
- PMID: 29100558
- PMCID: PMC6311987
- DOI: 10.1016/S2352-3026(17)30196-5
Immunohaematological complications in patients with sickle cell disease after haemopoietic progenitor cell transplantation: a prospective, single-centre, observational study
Abstract
Background: Haemopoietic progenitor cell (HPC) transplantation can cure sickle cell disease. Non-myeloablative conditioning typically results in donor-derived erythrocytes and stable mixed chimerism of recipient-derived and donor-derived leucocytes. Exposure to donor antigens from the HPC graft and new red cell antibodies induced by transfusion can lead to immunohaematological complications. We assessed the incidence of such complications among HPC transplant recipients with sickle cell disease.
Methods: The study population was all patients with sickle cell disease enrolled before March 31, 2015, in the three clinical trials of non-myeloablative HPC transplantation at the National Institutes of Health. We assessed formation of new red cell antibodies after transplantation and red cell incompatibility between donors and recipients.
Findings: 61 patients were enrolled, 42 were HLA matched and 19 were haploidentical. Nine (15%) had immunohaematological complications. Before HPC transplantation, three patients had antibodies incompatible with their donors. After HPC transplantation, new red cell antibodies were seen in six patients (11 alloantibodies and two autoantibodies), among whom three developed antibodies incompatible with donor or recipient red cells and three developed compatible antibodies. The clinical course of complications was highly variable, from no severe effects attributable to antibodies, to sustained reticulocytopenia, to near-fatal haemolysis. We found no significant correlation between immunohaematological complications and graft failure, graft rejection, or death.
Interpretation: Clinical effects ranged from seemingly not clinically important to potentially fatal. In patients with sickle cell disease, donor and recipient red cell phenotypes should be carefully assessed before transplantation to minimise and manage the risk of immunohaematological complications.
Funding: Intramural Research Program and National Institutes of Health.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declarations of interest
We declare no competing interests.
Figures
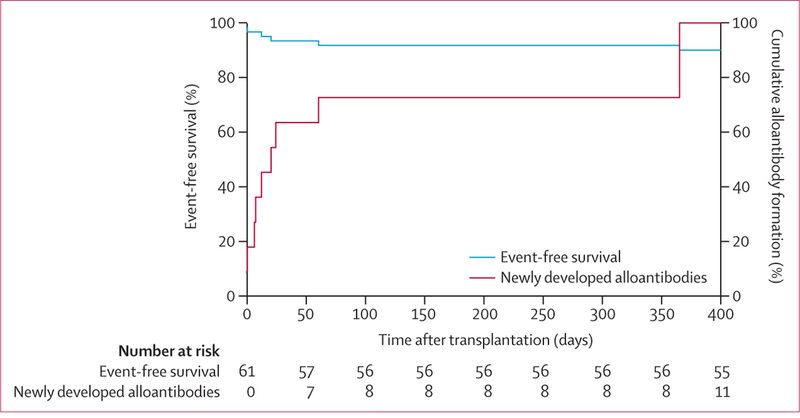
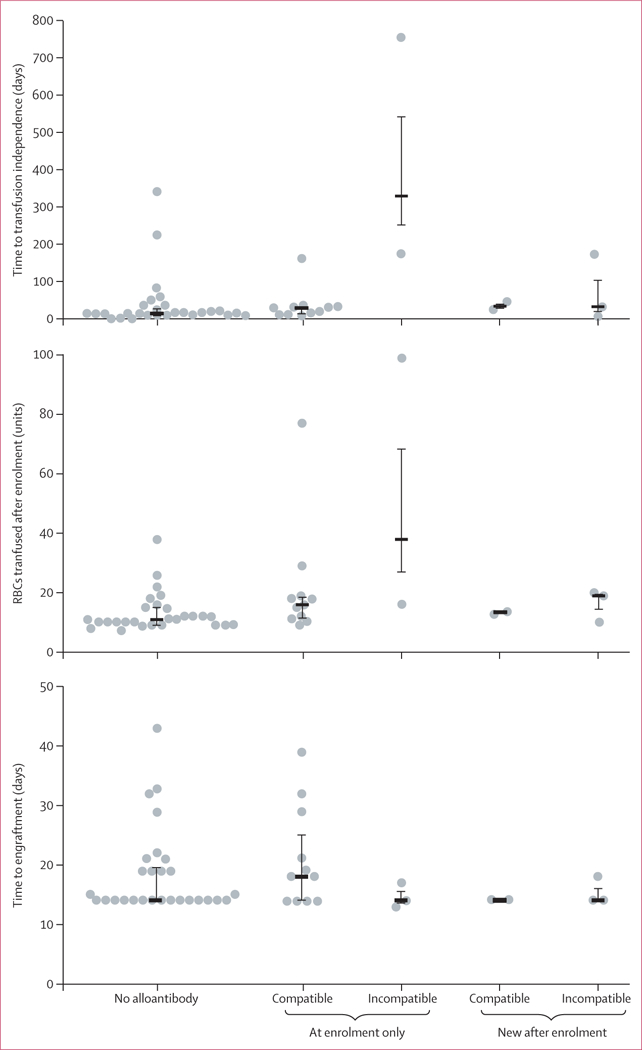
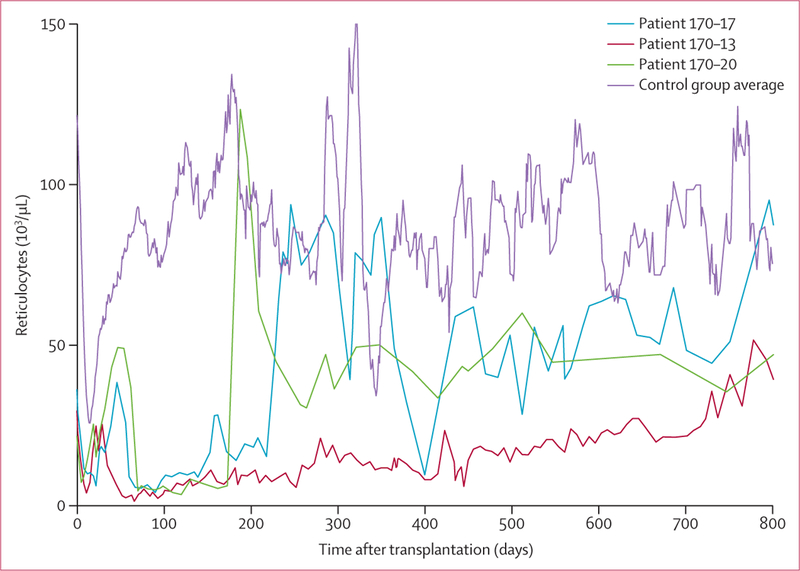
Comment in
-
Red blood cell alloimmunisation in patients with sickle cell disease.Lancet Haematol. 2017 Nov;4(11):e506-e507. doi: 10.1016/S2352-3026(17)30198-9. Lancet Haematol. 2017. PMID: 29100556 No abstract available.
Similar articles
-
Incidence and Role of Recipient-Specific Antibodies in Allogeneic Hematopoietic Cell Transplantation from Mismatched Related Donors.Transplant Cell Ther. 2024 Jan;30(1):99.e1-99.e10. doi: 10.1016/j.jtct.2023.10.015. Epub 2023 Oct 22. Transplant Cell Ther. 2024. PMID: 37875214
-
Uniform Graft-versus-Host Disease Prophylaxis using Post-Transplantation Cyclophosphamide, Methotrexate, and Cyclosporine following Peripheral Blood Hematopoietic Stem Cell Transplantation from Matched and Haploidentical Donors for Transfusion-Dependent Thalassemia: A Retrospective Report from the Bone Marrow Failure Working Group of Hunan Province, China.Transplant Cell Ther. 2024 Dec;30(12):1213.e1-1213.e12. doi: 10.1016/j.jtct.2024.08.022. Epub 2024 Sep 3. Transplant Cell Ther. 2024. PMID: 39236789
-
First-line allogeneic hematopoietic stem cell transplantation of HLA-matched sibling donors compared with first-line ciclosporin and/or antithymocyte or antilymphocyte globulin for acquired severe aplastic anemia.Cochrane Database Syst Rev. 2013 Jul 23;2013(7):CD006407. doi: 10.1002/14651858.CD006407.pub2. Cochrane Database Syst Rev. 2013. PMID: 23881658 Free PMC article.
-
Pilot Study of Donor-Engrafted Clonal Hematopoiesis Evolution and Clinical Outcomes in Allogeneic Hematopoietic Cell Transplantation Recipients Using a National Registry.Transplant Cell Ther. 2023 Oct;29(10):640.e1-640.e8. doi: 10.1016/j.jtct.2023.07.021. Epub 2023 Jul 28. Transplant Cell Ther. 2023. PMID: 37517612 Free PMC article.
-
Haploidentical Allogeneic Stem Cell Transplantation in Sickle Cell Disease: A Systematic Review and Meta-Analysis.Transplant Cell Ther. 2021 Dec;27(12):1004.e1-1004.e8. doi: 10.1016/j.jtct.2021.09.009. Epub 2021 Sep 17. Transplant Cell Ther. 2021. PMID: 34537420
Cited by
-
How we evaluate red blood cell compatibility and transfusion support for patients with sickle cell disease undergoing hematopoietic progenitor cell transplantation.Transfusion. 2018 Nov;58(11):2483-2489. doi: 10.1111/trf.14871. Epub 2018 Sep 28. Transfusion. 2018. PMID: 30403414 Free PMC article.
-
Therapeutic effect of autologous bone marrow stem cell mobilization combined with anti-infective therapy on moyamoya disease.Saudi J Biol Sci. 2020 Feb;27(2):676-681. doi: 10.1016/j.sjbs.2019.12.016. Epub 2019 Dec 19. Saudi J Biol Sci. 2020. PMID: 32210687 Free PMC article.
-
Transfusion Support in Hematopoietic Stem Cell Transplantation: A Contemporary Narrative Review.Clin Hematol Int. 2024 Mar 25;6(1):128-140. doi: 10.46989/001c.94135. eCollection 2024. Clin Hematol Int. 2024. PMID: 38817704 Free PMC article. Review.
-
CD47 regulates antigen modulation and red blood cell clearance following an incompatible transfusion.Front Immunol. 2025 Apr 4;16:1548548. doi: 10.3389/fimmu.2025.1548548. eCollection 2025. Front Immunol. 2025. PMID: 40255405 Free PMC article.
-
Molecular immunohaematology round table discussions at the AABB Annual Meeting, Orlando 2016.Blood Transfus. 2018 Sep;16(5):447-456. doi: 10.2450/2018.0260-17. Epub 2018 Feb 14. Blood Transfus. 2018. PMID: 29517973 Free PMC article. No abstract available.
References
-
- Johnson FL, Look AT, Gockerman J, Ruggiero MR, Dalla-Pozza L, Billings FT 3rd. Bone-marrow transplantation in a patient with sickle-cell anemia. N Engl J Med 1984; 311: 780–83. - PubMed
-
- Vermylen C, Cornu G, Ferster A, et al. Haematopoietic stem cell transplantation for sickle cell anaemia: the first 50 patients transplanted in Belgium. Bone Marrow Transplant 1998; 22: 1–6. - PubMed
-
- Walters MC, Patience M, Leisenring W, et al. Stable mixed hematopoietic chimerism after bone marrow transplantation for sickle cell anemia. Biol Blood Marrow Transplant 2001; 7: 665–73. - PubMed
-
- van Besien K, Bartholomew A, Stock W, et al. Fludarabine-based conditioning for allogeneic transplantation in adults with sickle cell disease. Bone Marrow Transplant 2000; 26: 445^−9. - PubMed
-
- Iannone R, Casella JF, Fuchs EJ, et al. Results of minimally toxic nonmyeloablative transplantation in patients with sickle cell anemia and beta-thalassemia. Biol Blood Marrow Transplant 2003; 9: 519–28. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
