

Cardiotoxicity of immune checkpoint inhibitors
- PMID: 29104763
- PMCID: PMC5663252
- DOI: 10.1136/esmoopen-2017-000247
Cardiotoxicity of immune checkpoint inhibitors
Abstract
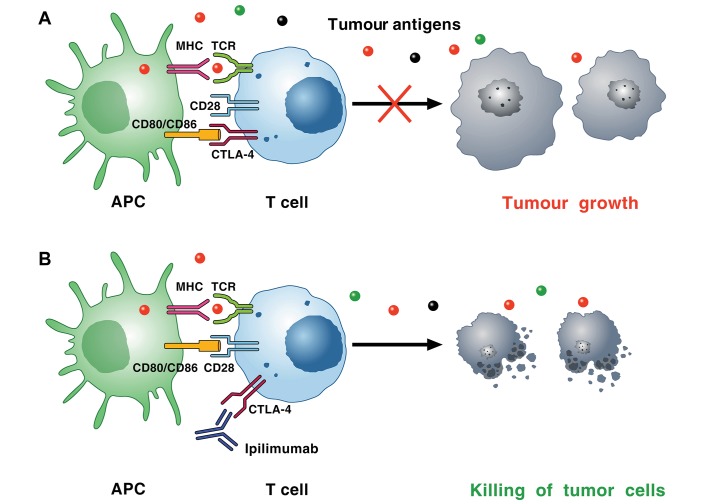
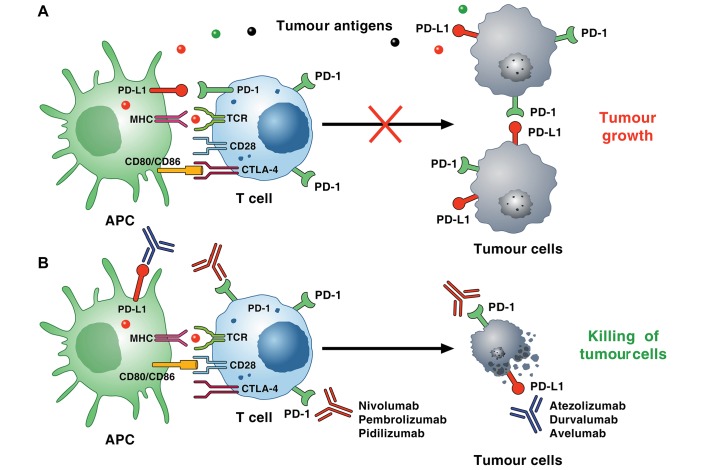
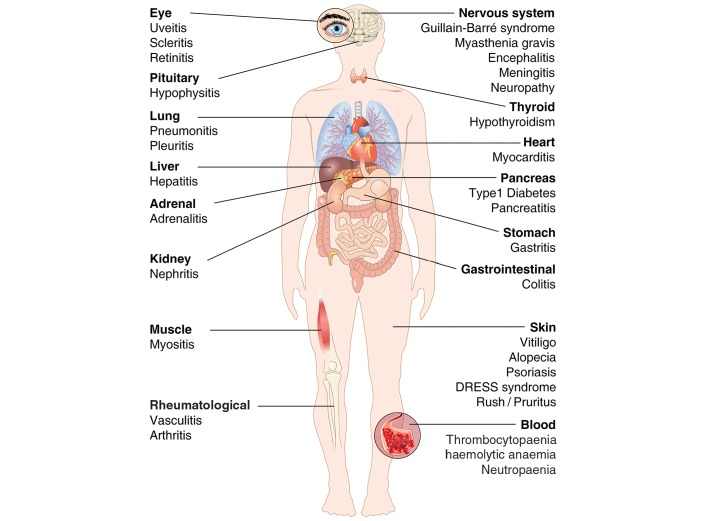
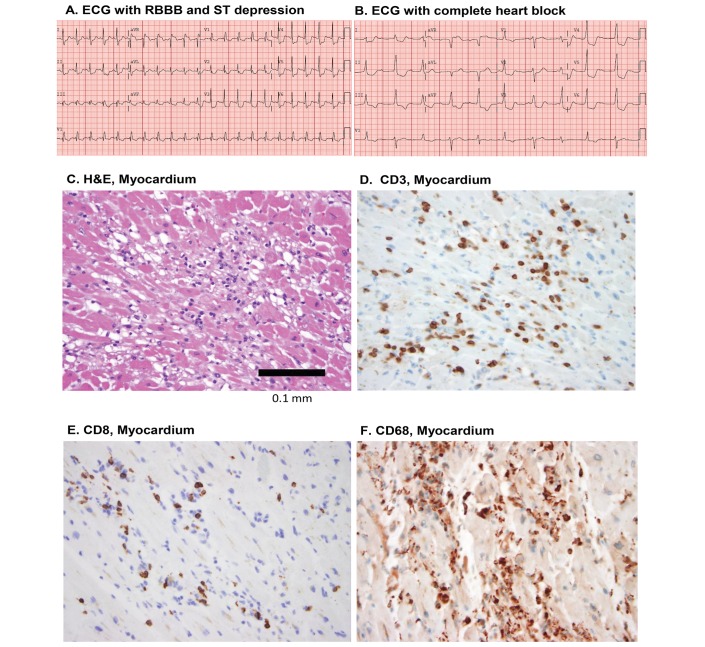
Cardiac toxicity after conventional antineoplastic drugs (eg, anthracyclines) has historically been a relevant issue. In addition, targeted therapies and biological molecules can also induce cardiotoxicity. Immune checkpoint inhibitors are a novel class of anticancer drugs, distinct from targeted or tumour type-specific therapies. Cancer immunotherapy with immune checkpoint blockers (ie, monoclonal antibodies targeting cytotoxic T lymphocyte-associated antigen 4 (CTLA-4), programmed cell death 1 (PD-1) and its ligand (PD-L1)) has revolutionised the management of a wide variety of malignancies endowed with poor prognosis. These inhibitors unleash antitumour immunity, mediate cancer regression and improve the survival in a percentage of patients with different types of malignancies, but can also produce a wide spectrum of immune-related adverse events. Interestingly, PD-1 and PD-L1 are expressed in rodent and human cardiomyocytes, and early animal studies have demonstrated that CTLA-4 and PD-1 deletion can cause autoimmune myocarditis. Cardiac toxicity has largely been underestimated in recent reviews of toxicity of checkpoint inhibitors, but during the last years several cases of myocarditis and fatal heart failure have been reported in patients treated with checkpoint inhibitors alone and in combination. Here we describe the mechanisms of the most prominent checkpoint inhibitors, specifically ipilimumab (anti-CTLA-4, the godfather of checkpoint inhibitors) patient and monoclonal antibodies targeting PD-1 (eg, nivolumab, pembrolizumab) and PD-L1 (eg, atezolizumab). We also discuss what is known and what needs to be done about cardiotoxicity of checkpoint inhibitors in patients with cancer. Severe cardiovascular effects associated with checkpoint blockade introduce important issues for oncologists, cardiologists and immunologists.
Keywords: CTLA-4; PD-1; PD-L1; cancer; cardiotoxicity; immune checkpoints; melanoma; myocarditis.
Conflict of interest statement
Competing interests: CGT received travel support from Alere.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
