

S-sulfocysteine/NMDA receptor-dependent signaling underlies neurodegeneration in molybdenum cofactor deficiency
- PMID: 29106383
- PMCID: PMC5707142
- DOI: 10.1172/JCI89885
S-sulfocysteine/NMDA receptor-dependent signaling underlies neurodegeneration in molybdenum cofactor deficiency
Abstract
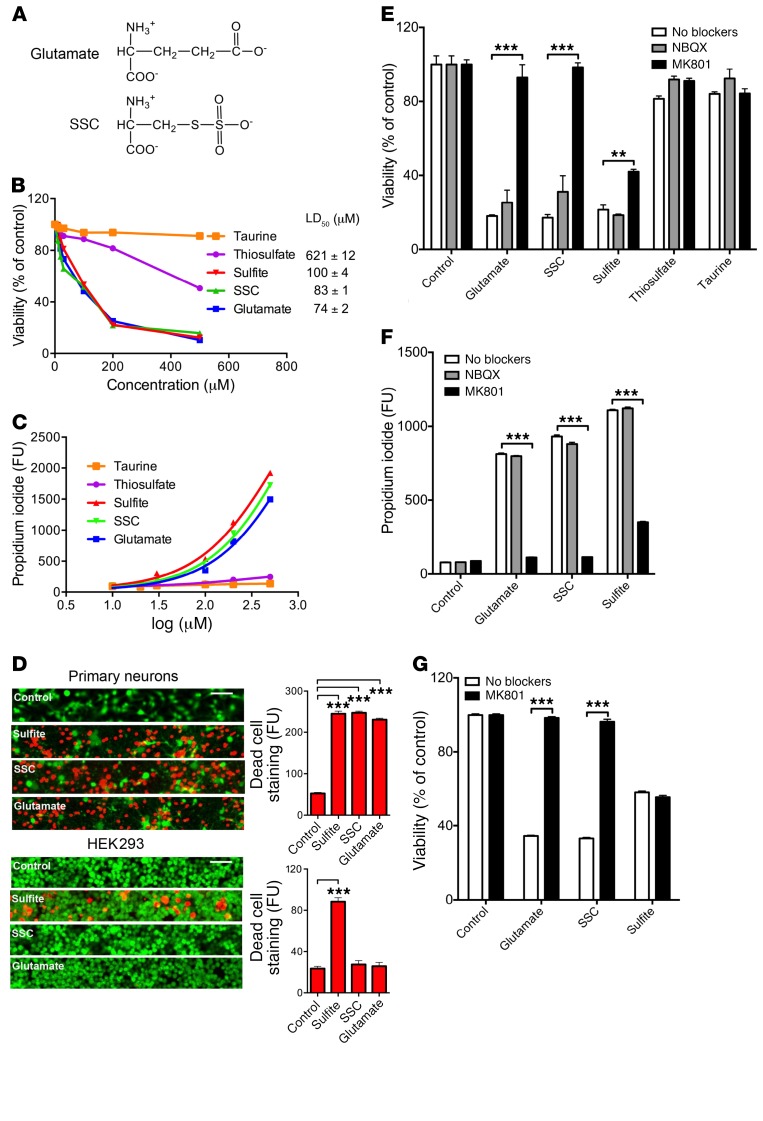
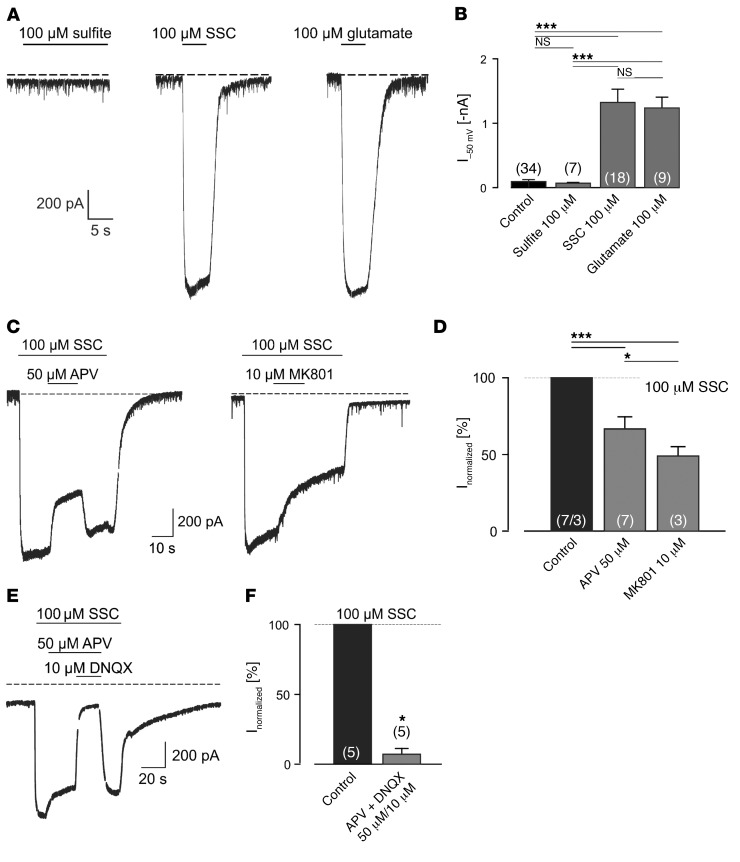
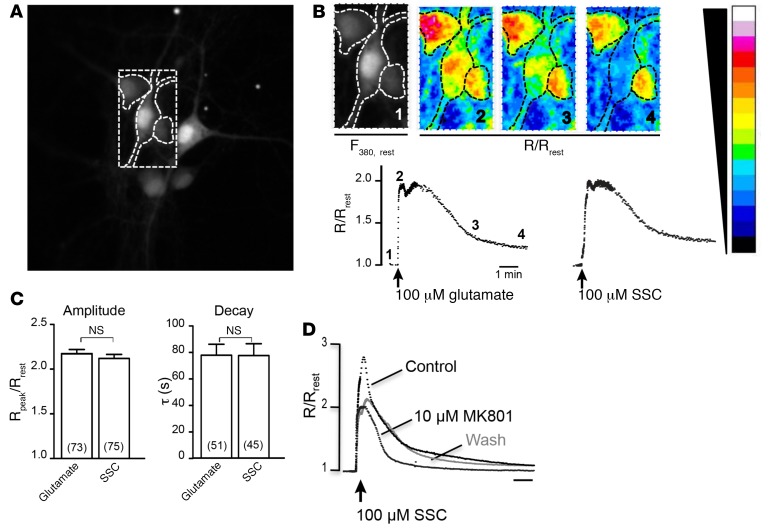
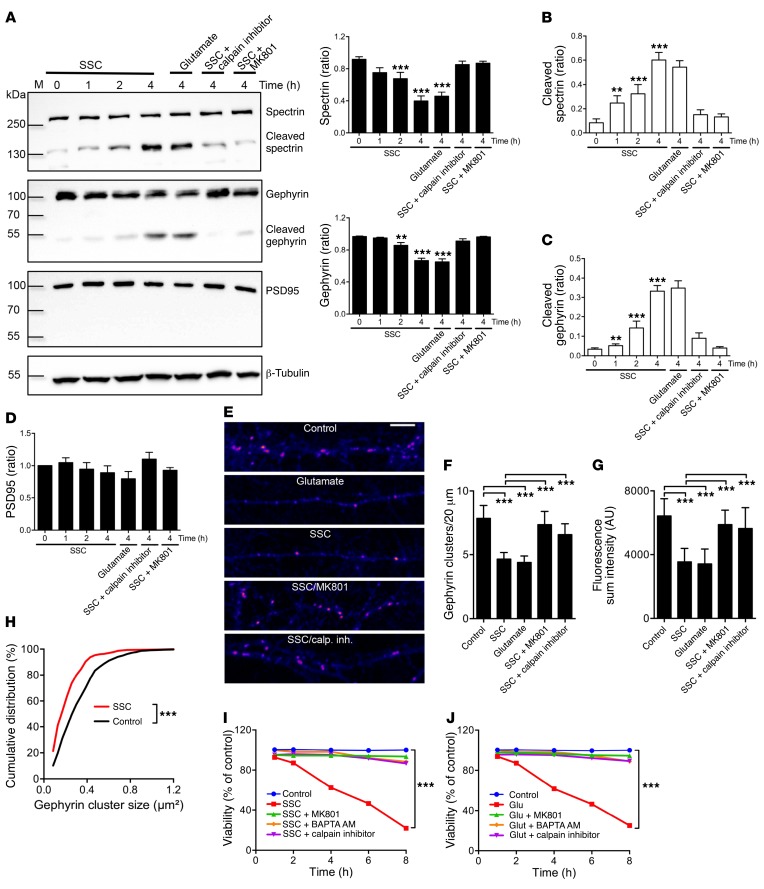
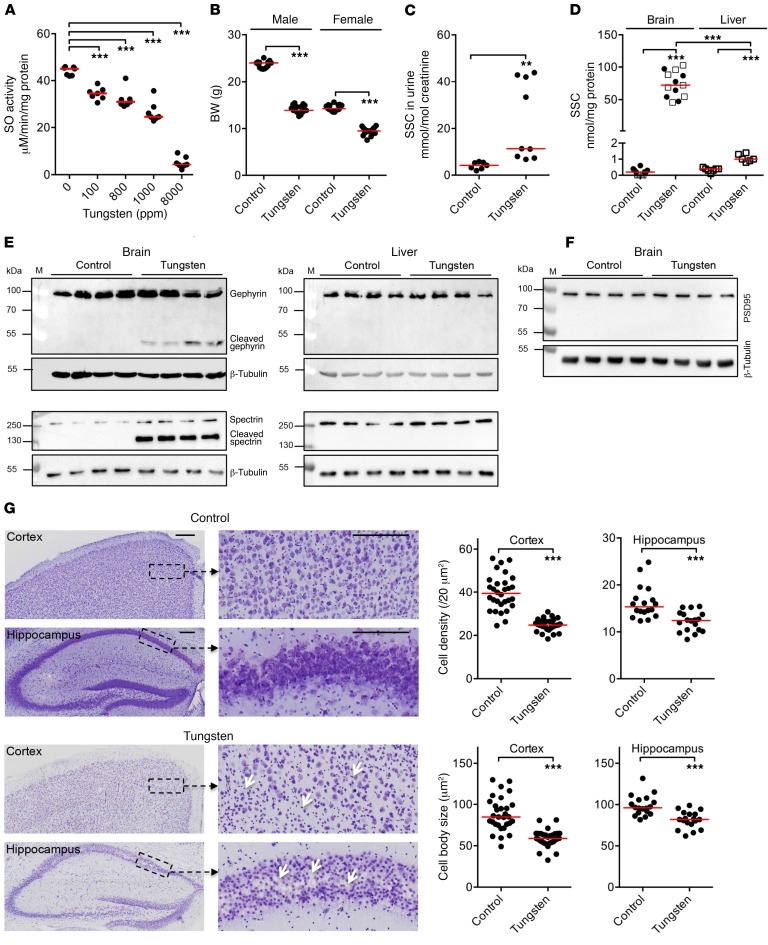
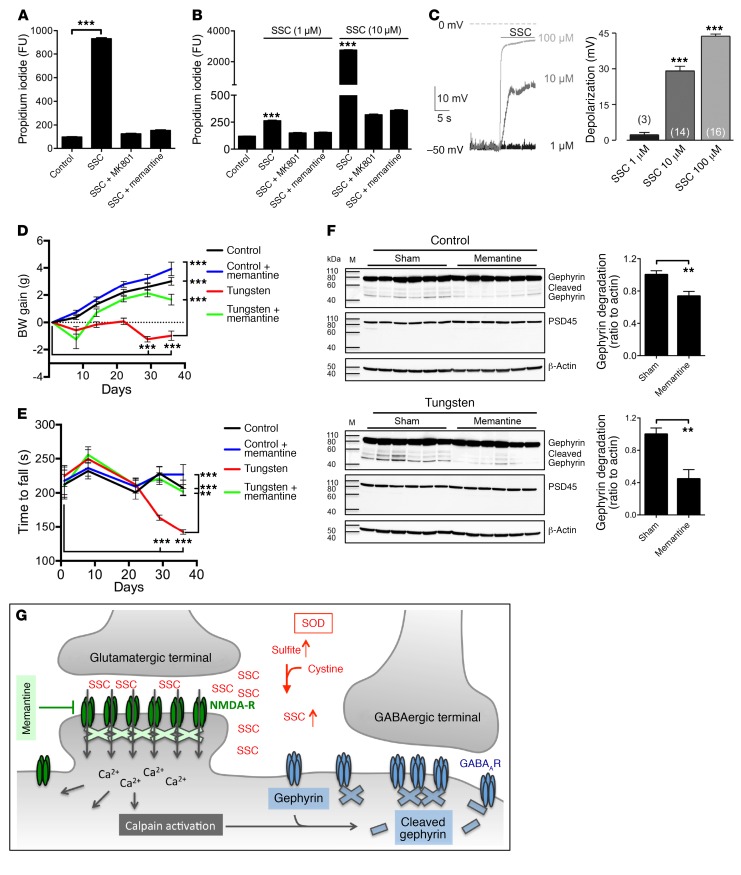
Molybdenum cofactor deficiency (MoCD) is an autosomal recessive inborn error of metabolism characterized by neurodegeneration and death in early childhood. The rapid and progressive neurodegeneration in MoCD presents a major clinical challenge and may relate to the poor understanding of the molecular mechanisms involved. Recently, we reported that treating patients with cyclic pyranopterin monophosphate (cPMP) is a successful therapy for a subset of infants with MoCD and prevents irreversible brain damage. Here, we studied S-sulfocysteine (SSC), a structural analog of glutamate that accumulates in the plasma and urine of patients with MoCD, and demonstrated that it acts as an N-methyl D-aspartate receptor (NMDA-R) agonist, leading to calcium influx and downstream cell signaling events and neurotoxicity. SSC treatment activated the protease calpain, and calpain-dependent degradation of the inhibitory synaptic protein gephyrin subsequently exacerbated SSC-mediated excitotoxicity and promoted loss of GABAergic synapses. Pharmacological blockade of NMDA-R, calcium influx, or calpain activity abolished SSC and glutamate neurotoxicity in primary murine neurons. Finally, the NMDA-R antagonist memantine was protective against the manifestation of symptoms in a tungstate-induced MoCD mouse model. These findings demonstrate that SSC drives excitotoxic neurodegeneration in MoCD and introduce NMDA-R antagonists as potential therapeutics for this fatal disease.
Keywords: Neurodegeneration; Neuroscience.
Conflict of interest statement
Figures
References
-
- Johnson JL, Waud WR, Rajagopalan KV, Duran M, Beemer FA, Wadman SK. Inborn errors of molybdenum metabolism: combined deficiencies of sulfite oxidase and xanthine dehydrogenase in a patient lacking the molybdenum cofactor. Proc Natl Acad Sci U S A. 1980;77(6):3715–3719. doi: 10.1073/pnas.77.6.3715. - DOI - PMC - PubMed
-
- Johnson JL, Duran M. Molybdenum cofactor deficiency and isolated sulfite oxidase deficiency. In: Scriver C, Beaudet A, Sly W, Valle D, eds. The metabolic and molecular bases of inherited disease. New York, NY: McGraw-Hill; 2001:3163–3177.
Publication types
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
