

ProteomicsDB
- PMID: 29106664
- PMCID: PMC5753189
- DOI: 10.1093/nar/gkx1029
ProteomicsDB
Abstract
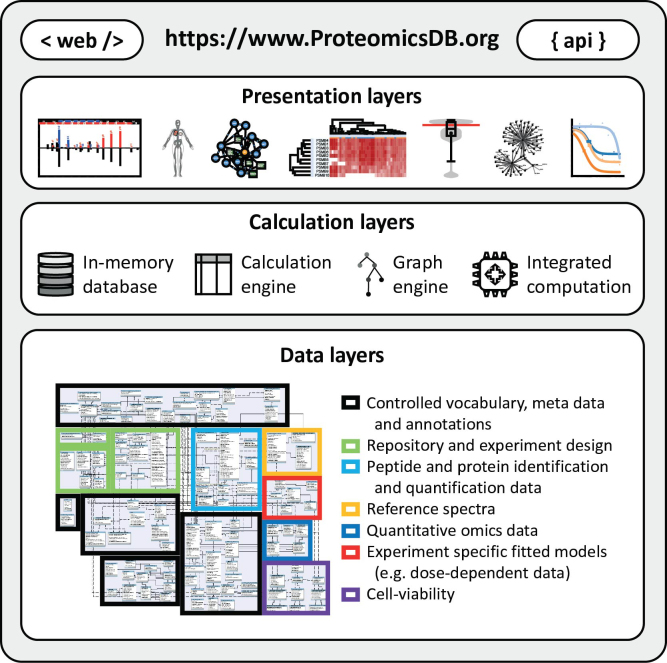
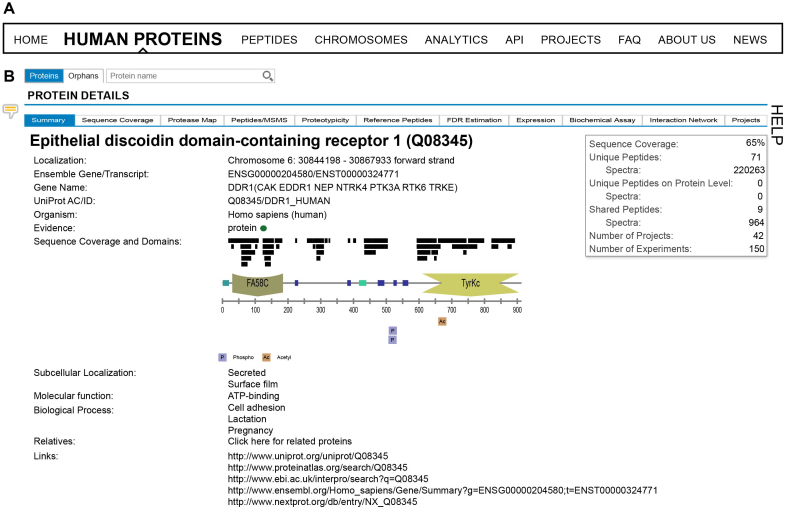
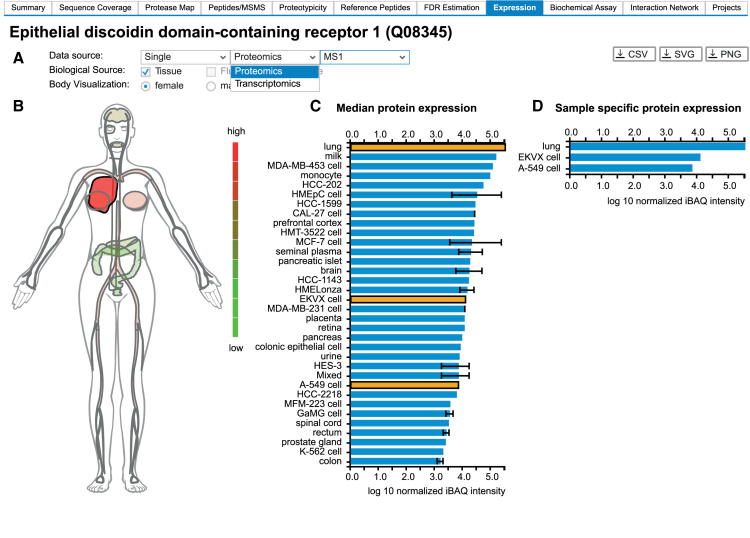
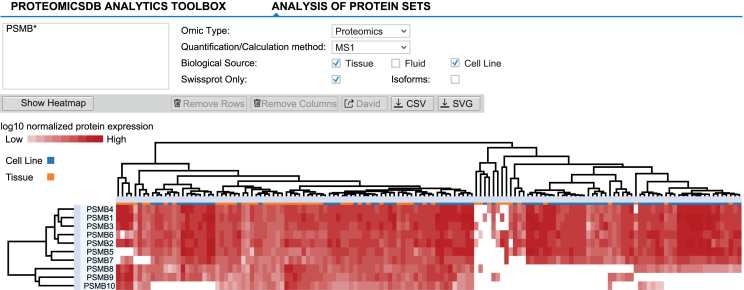
ProteomicsDB (https://www.ProteomicsDB.org) is a protein-centric in-memory database for the exploration of large collections of quantitative mass spectrometry-based proteomics data. ProteomicsDB was first released in 2014 to enable the interactive exploration of the first draft of the human proteome. To date, it contains quantitative data from 78 projects totalling over 19k LC-MS/MS experiments. A standardized analysis pipeline enables comparisons between multiple datasets to facilitate the exploration of protein expression across hundreds of tissues, body fluids and cell lines. We recently extended the data model to enable the storage and integrated visualization of other quantitative omics data. This includes transcriptomics data from e.g. NCBI GEO, protein-protein interaction information from STRING, functional annotations from KEGG, drug-sensitivity/selectivity data from several public sources and reference mass spectra from the ProteomeTools project. The extended functionality transforms ProteomicsDB into a multi-purpose resource connecting quantification and meta-data for each protein. The rich user interface helps researchers to navigate all data sources in either a protein-centric or multi-protein-centric manner. Several options are available to download data manually, while our application programming interface enables accessing quantitative data systematically.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
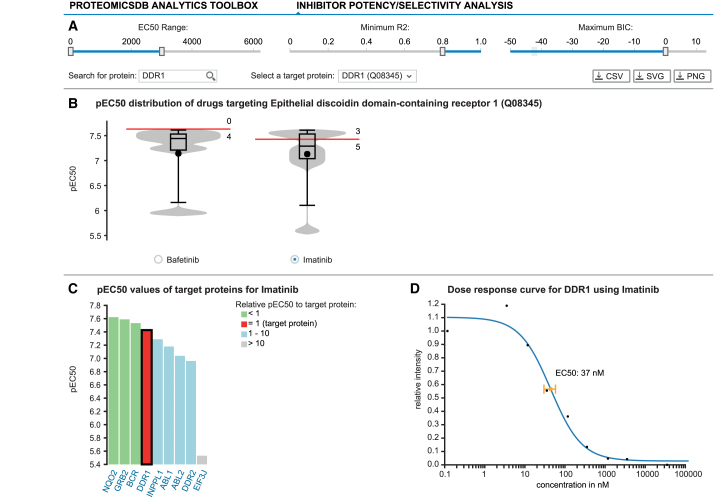
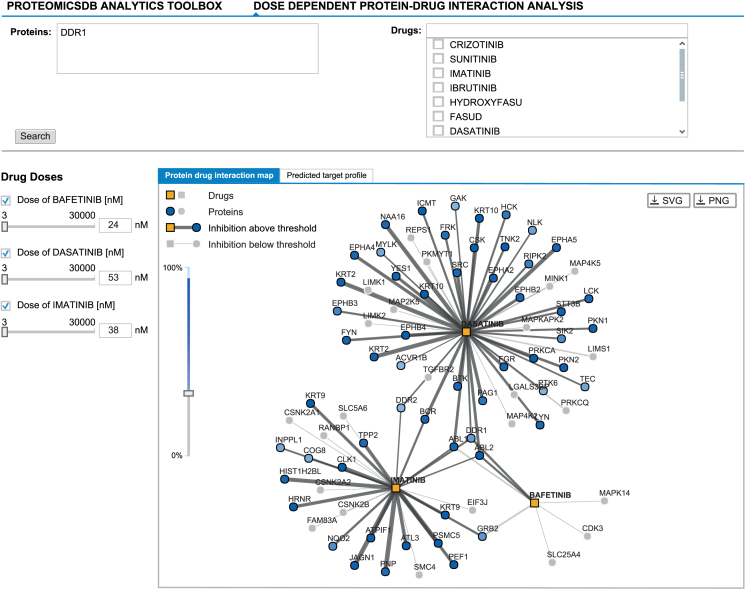
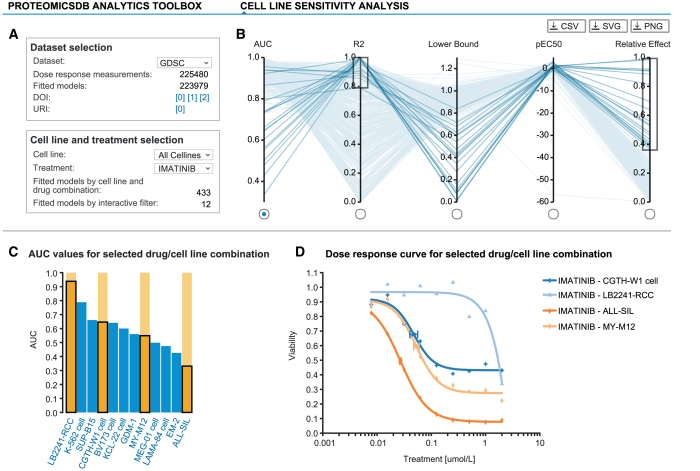
References
-
- Aebersold R., Mann M.. Mass spectrometry-based proteomics. Nature. 2003; 422:198–207. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
