

Epigenetic memory via concordant DNA methylation is inversely correlated to developmental potential of mammalian cells
- PMID: 29107996
- PMCID: PMC5690686
- DOI: 10.1371/journal.pgen.1007060
Epigenetic memory via concordant DNA methylation is inversely correlated to developmental potential of mammalian cells
Abstract
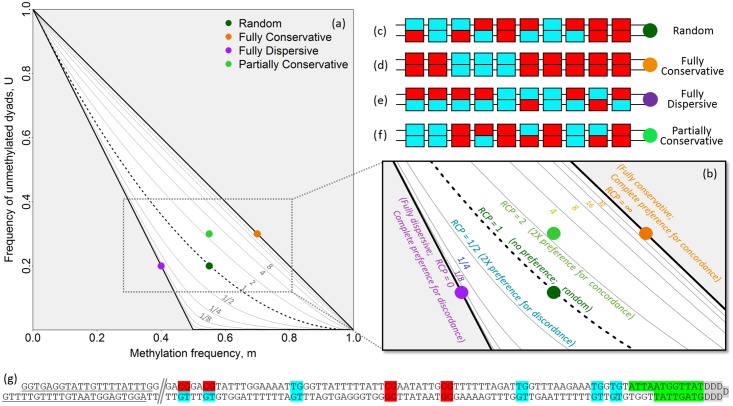
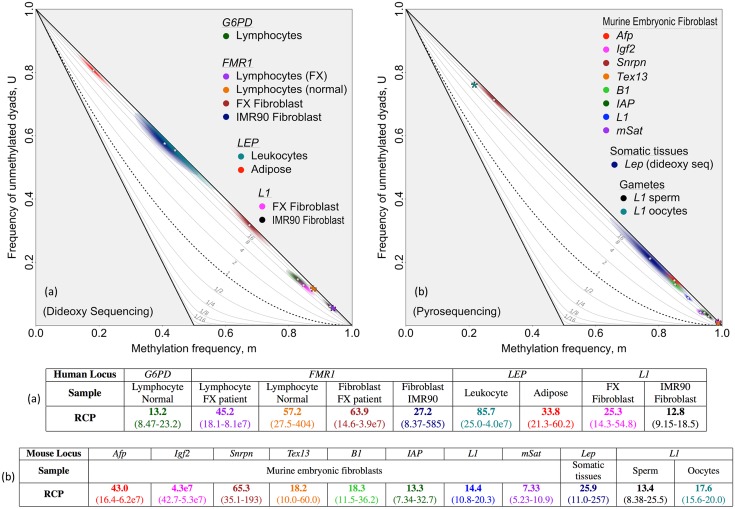
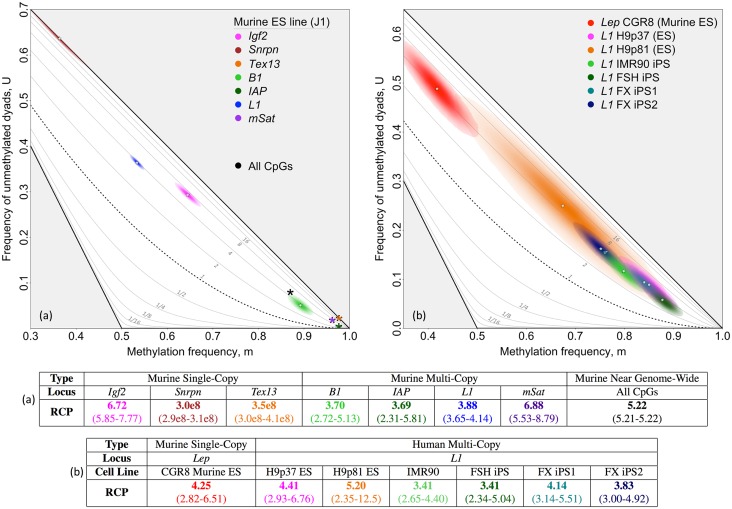
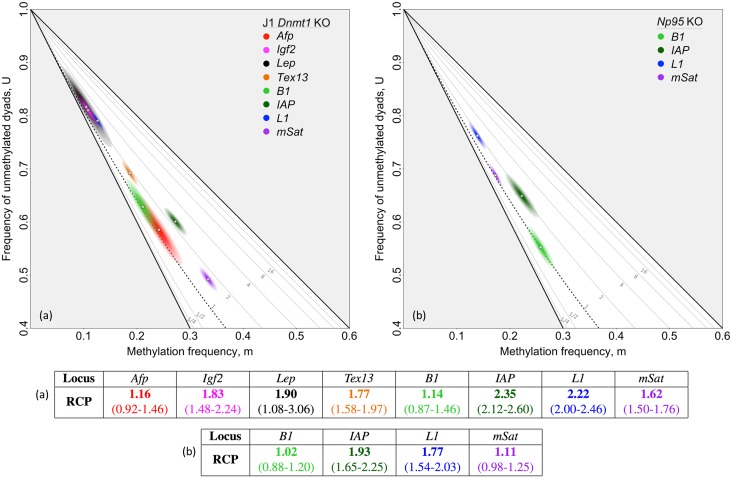
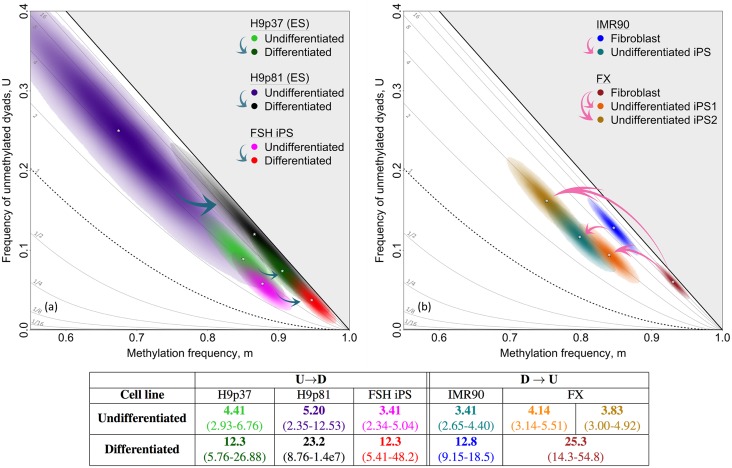
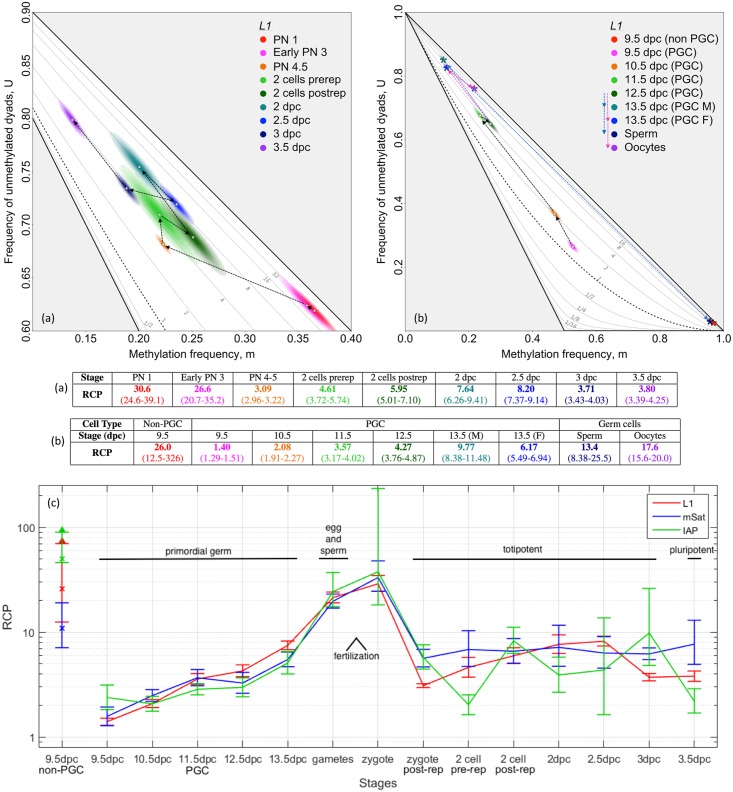
In storing and transmitting epigenetic information, organisms must balance the need to maintain information about past conditions with the capacity to respond to information in their current and future environments. Some of this information is encoded by DNA methylation, which can be transmitted with variable fidelity from parent to daughter strand. High fidelity confers strong pattern matching between the strands of individual DNA molecules and thus pattern stability over rounds of DNA replication; lower fidelity confers reduced pattern matching, and thus greater flexibility. Here, we present a new conceptual framework, Ratio of Concordance Preference (RCP), that uses double-stranded methylation data to quantify the flexibility and stability of the system that gave rise to a given set of patterns. We find that differentiated mammalian cells operate with high DNA methylation stability, consistent with earlier reports. Stem cells in culture and in embryos, in contrast, operate with reduced, albeit significant, methylation stability. We conclude that preference for concordant DNA methylation is a consistent mode of information transfer, and thus provides epigenetic stability across cell divisions, even in stem cells and those undergoing developmental transitions. Broader application of our RCP framework will permit comparison of epigenetic-information systems across cells, developmental stages, and organisms whose methylation machineries differ substantially or are not yet well understood.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Feinberg AP, Vogelstein B, et al. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301(5895):89–92. doi: 10.1038/301089a0 - DOI - PubMed
-
- Baylin SB, Jones PA. A decade of exploring the cancer epigenome—biological and translational implications. Nature Reviews Cancer. 2011;11(10):726–734. doi: 10.1038/nrc3130 - DOI - PMC - PubMed
-
- Ohi Y, Qin H, Hong C, Blouin L, Polo JM, Guo T, et al. Incomplete DNA methylation underlies a transcriptional memory of somatic cells in human iPS cells. Nature cell biology. 2011;13(5):541–549. doi: 10.1038/ncb2239 - DOI - PMC - PubMed
-
- Monk M, Boubelik M, Lehnert S. Temporal and regional changes in DNA methylation in the embryonic, extraembryonic and germ cell lineages during mouse embryo development. Development. 1987;99(3):371–382. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
