

Mapping and phasing of structural variation in patient genomes using nanopore sequencing
- PMID: 29109544
- PMCID: PMC5673902
- DOI: 10.1038/s41467-017-01343-4
Mapping and phasing of structural variation in patient genomes using nanopore sequencing
Abstract
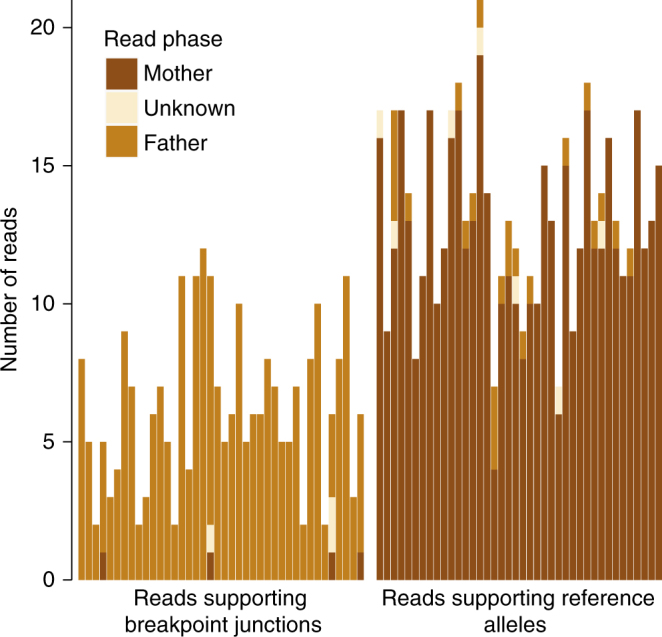
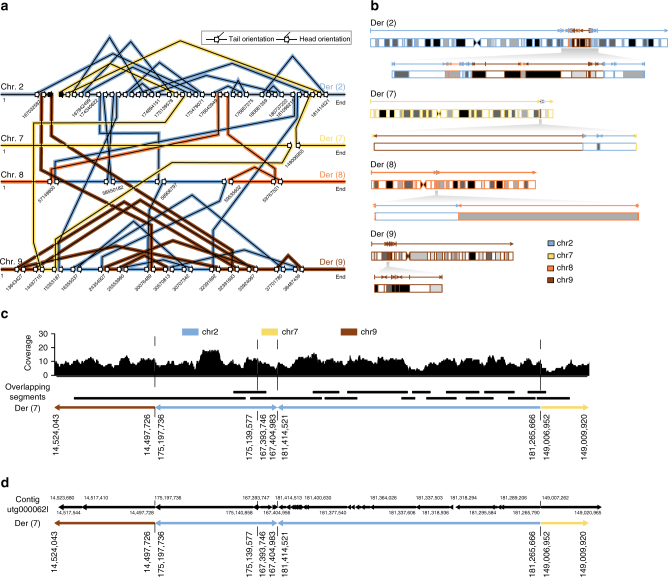
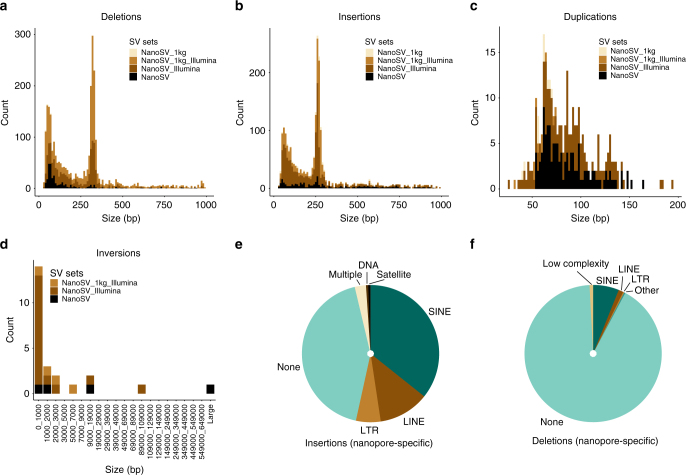
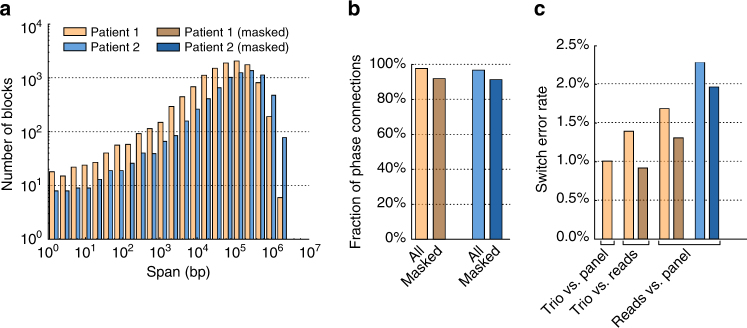
Despite improvements in genomics technology, the detection of structural variants (SVs) from short-read sequencing still poses challenges, particularly for complex variation. Here we analyse the genomes of two patients with congenital abnormalities using the MinION nanopore sequencer and a novel computational pipeline-NanoSV. We demonstrate that nanopore long reads are superior to short reads with regard to detection of de novo chromothripsis rearrangements. The long reads also enable efficient phasing of genetic variations, which we leveraged to determine the parental origin of all de novo chromothripsis breakpoints and to resolve the structure of these complex rearrangements. Additionally, genome-wide surveillance of inherited SVs reveals novel variants, missed in short-read data sets, a large proportion of which are retrotransposon insertions. We provide a first exploration of patient genome sequencing with a nanopore sequencer and demonstrate the value of long-read sequencing in mapping and phasing of SVs for both clinical and research applications.
Conflict of interest statement
W.P.K. and J.d.R. have received financial compensation for travel and accommodation expenses to speak at an Oxford Nanopore Technologies-organized meeting. The remaining authors declare no competing financial interests.
Figures

References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
