

Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems
- PMID: 29111720
- PMCID: PMC5726918
- DOI: 10.1021/acs.jctc.7b00875
Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems
Erratum in
-
Correction to Constant-pH Molecular Dynamics Simulations for Large Biomolecular Systems.J Chem Theory Comput. 2018 Dec 11;14(12):6748-6749. doi: 10.1021/acs.jctc.8b01075. Epub 2018 Nov 27. J Chem Theory Comput. 2018. PMID: 30480445 Free PMC article. No abstract available.
Abstract
An increasingly important endeavor is to develop computational strategies that enable molecular dynamics (MD) simulations of biomolecular systems with spontaneous changes in protonation states under conditions of constant pH. The present work describes our efforts to implement the powerful constant-pH MD simulation method, based on a hybrid nonequilibrium MD/Monte Carlo (neMD/MC) technique within the highly scalable program NAMD. The constant-pH hybrid neMD/MC method has several appealing features; it samples the correct semigrand canonical ensemble rigorously, the computational cost increases linearly with the number of titratable sites, and it is applicable to explicit solvent simulations. The present implementation of the constant-pH hybrid neMD/MC in NAMD is designed to handle a wide range of biomolecular systems with no constraints on the choice of force field. Furthermore, the sampling efficiency can be adaptively improved on-the-fly by adjusting algorithmic parameters during the simulation. Illustrative examples emphasizing medium- and large-scale applications on next-generation supercomputing architectures are provided.
Figures
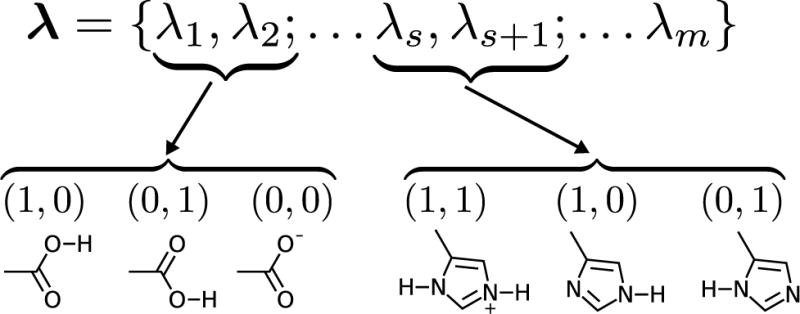

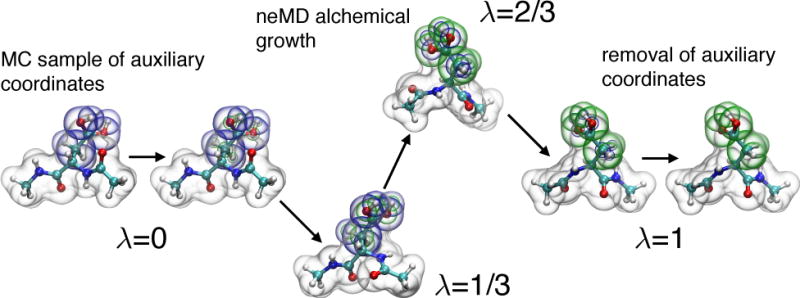
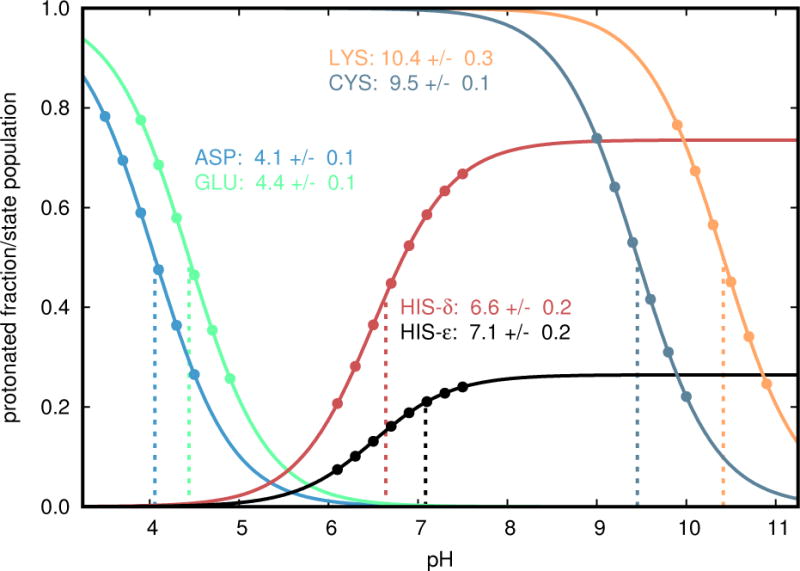
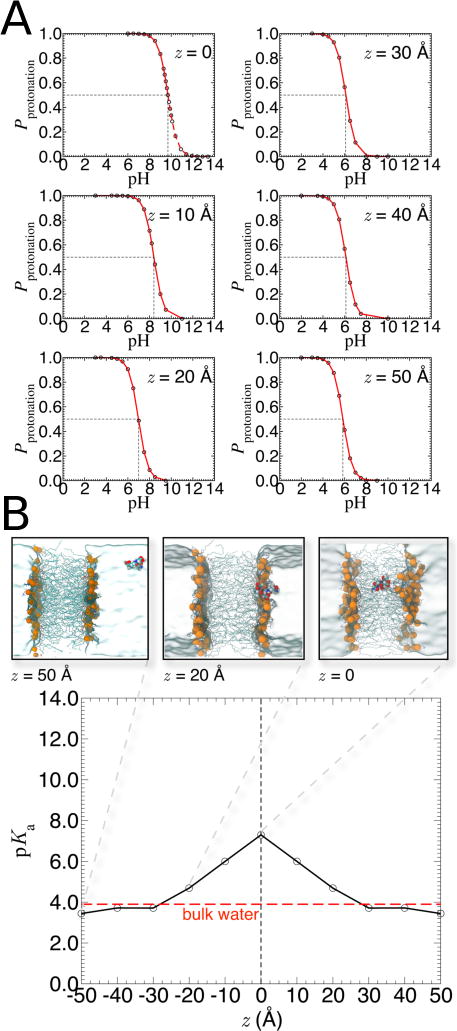
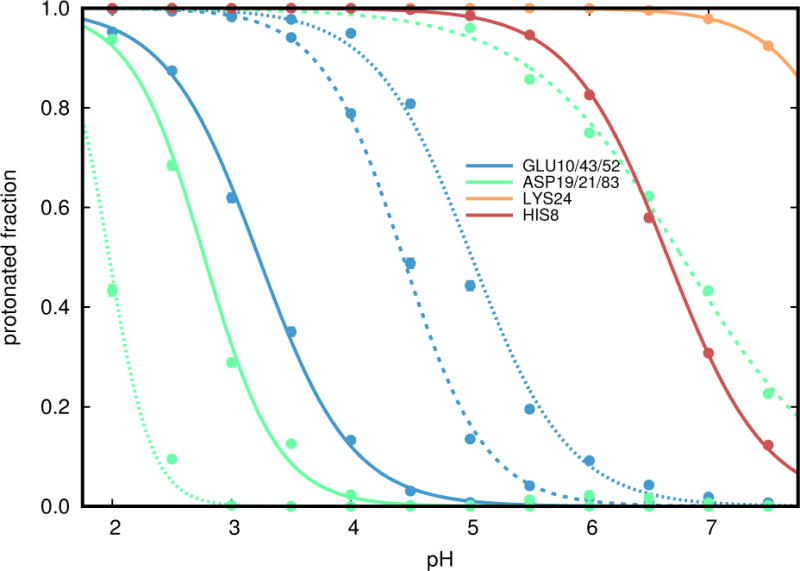
References
-
- Karplus M, McCammon JA. Molecular Dynamics Simulations of Biomolecules. Nature Struct Biol. 2002;9:646–652. - PubMed
-
- Dror RO, Dirks RM, Grossman JP, Xu H, Shaw DE. Biomolecular Simulation: A Computational Microscope for Molecular Biology. Annu Rev Biophys. 2012;41:429–452. - PubMed
-
- Nelson DL, Cox MM. Lehninger Principles of Biochemistry. 4th. W. H. Freeman; 2005.
-
- Webb BA, Chimenti M, Jacobson MP, Barber DL. Dysregulated pH: A Perfect Storm for Cancer Progression. Nat Rev Cancer. 2011;11:671–677. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources