

Review
doi: 10.1021/acs.chemrev.7b00205.
Epub 2017 Nov 7.
Chemical Biology of H2S Signaling through Persulfidation
Affiliations
- PMID: 29112440
- PMCID: PMC6029264
- DOI: 10.1021/acs.chemrev.7b00205
Item in Clipboard
Review
Chemical Biology of H2S Signaling through Persulfidation
Chem Rev.
.
Abstract
Signaling by H2S is proposed to occur via persulfidation, a posttranslational modification of cysteine residues (RSH) to persulfides (RSSH). Persulfidation provides a framework for understanding the physiological and pharmacological effects of H2S. Due to the inherent instability of persulfides, their chemistry is understudied. In this review, we discuss the biologically relevant chemistry of H2S and the enzymatic routes for its production and oxidation. We cover the chemical biology of persulfides and the chemical probes for detecting them. We conclude by discussing the roles ascribed to protein persulfidation in cell signaling pathways.
Conflict of interest statement
The authors declare no competing financial interest.
Figures
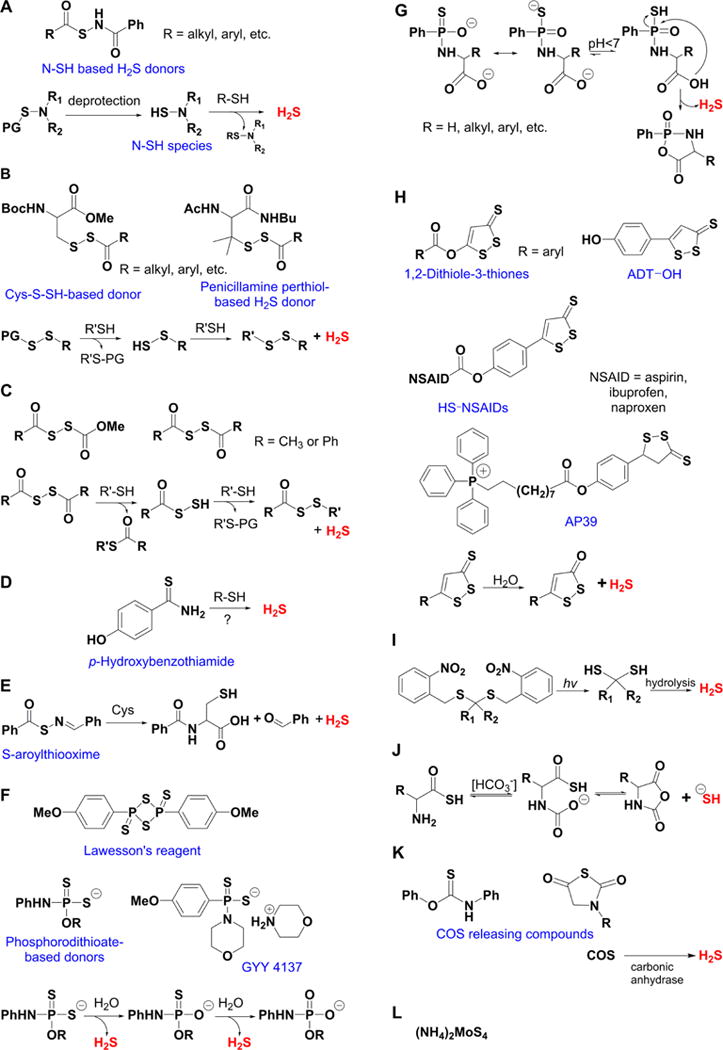
Overview of different classes of H2S donors. (A) Compounds based on the N-mercapto template (N-SH species) and the proposed mechanism for H2S release. PG, protective group. (B) Perthiol-based compounds and proposed thiol-dependent mechanism of H2S release. (C) Dithioperoxyanhydrides can also serve as H2S donors upon reaction with thiols. (D) Arylthioamides release H2S in the presence of thiols via an uncharacterized mechanism. (E) S-Aroylthiooximes release H2S in the presence of aminothiols. (F) Chemical structures of Lawesson’s reagent and its derivative, GYY4137, the most widely used H2S donor, and the proposed mechanism for H2S release from GYY4137. (G) Phosphorothioate-based H2S donors that release H2S in a pH-dependent manner. (H) Another widely used class of molecules is 1,2-dithiole-3-thiones. They can be coupled to nonsteroid antiinflammatory drugs (NSAID) such as aspirin, ibuprofen, or naproxen, or to triphenylphsophonium group (AP39) which directs them to mitochondria. This class of molecules is believed to release H2S via hydrolysis. (I) Example of photo cleavable gem-dithiol based H2S donors, which undergo hydrolysis to release H2S. (J) Thioamino acids release H2S in reactions with bicarbonate. (K) COS, released by COS donors, forms H2S in the presence of carbonic anhydrase. (L) Ammonium tetrathiomolibdate is shown to act as H2S donor in vivo.
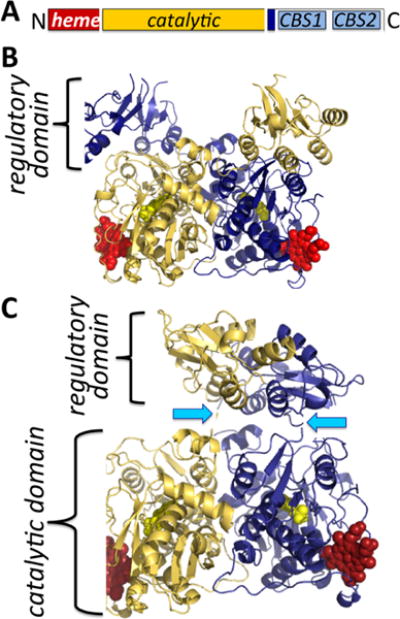
Organization and structure of human CBS. (A) CBS is a modular protein with regulatory domains at its N- and C-termini. The C-terminal domain comprises a tandem repeat of two CBS domains, CBS1 and CBS2. The structures of human CBS in the absence (PDB: 4L27) (B) and presence (PDB: 4PCU) (C) of AdoMet show that a large conformational rearrangement accompanies the transition from the basal to the activated state. The protomers are shown in blue and yellow, respectively, the heme (red) and PLP (yellow) are in sphere representation, and the blue arrows point to the intervening linker region between the catalytic core and the C-terminal domain.
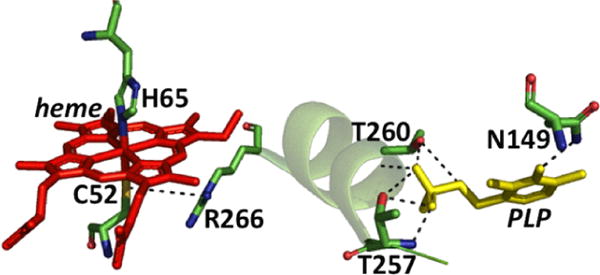
Close up of the CBS structure. The interactions between the Cys52 heme ligand and Arg266 at one end of the α-helix and between Thr257 and Thr260 and the phosphate group of PLP at the other are shown. Asn149 hydrogen bonds with the C4 oxygen in PLP.
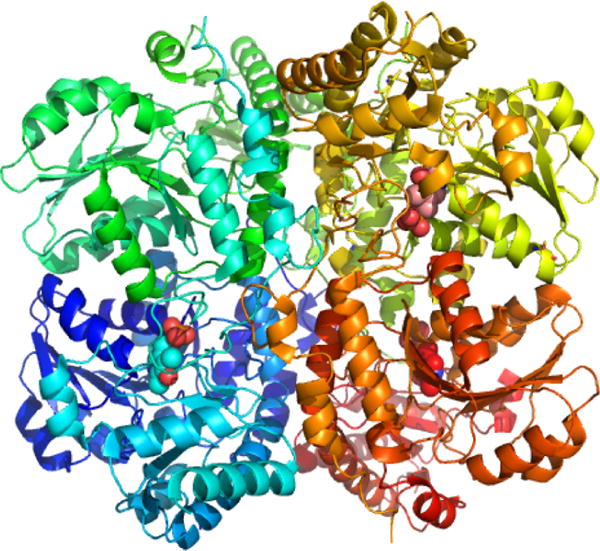
Structure of human CSE (PDB: 2NMP). Each of the four monomers is shown in a different color, and the three PLPs visible in the structure are in sphere representation.
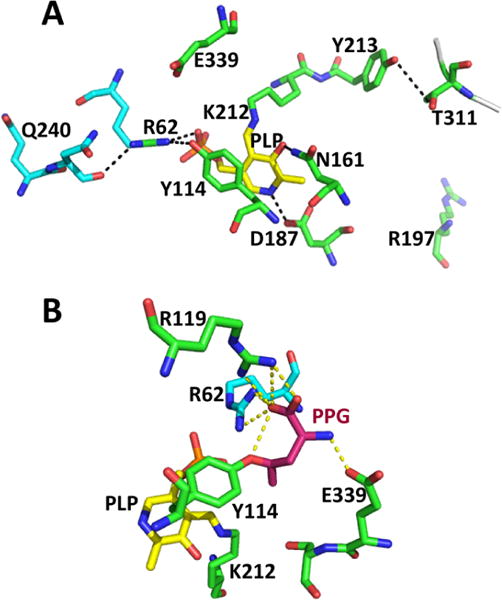
Close-up of the active site structure of human CSE. (A). Interactions between PLP (yellow) and residues donated by the two subunits (in green and cyan) are highlighted (PDB: 2NMP). (B). Structure of PPG-inactivated CSE (PDB: 3COG). The coloring is the same is in panel A. PPG is covalently linked to Tyr114 and is shown in pink.
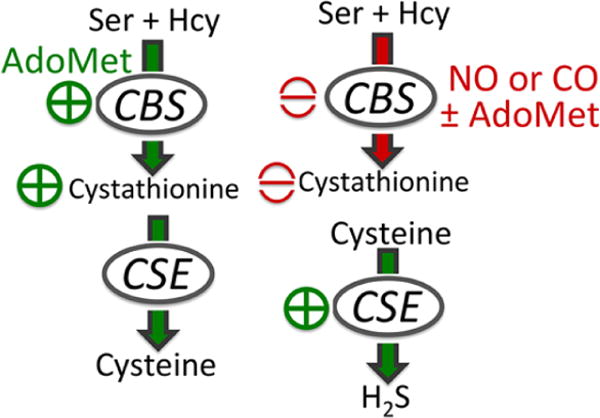
Heme-regulated switching in the transsulfuration pathway from cysteine (left) to H2S (right) synthesis.
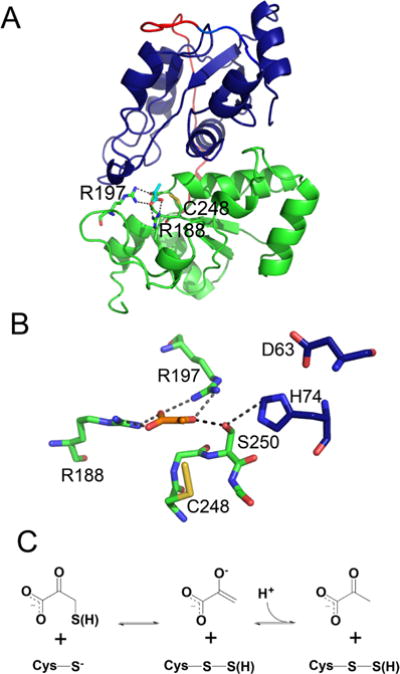
Structure and mechanism of human MST. (A) The N and C-terminal domains of human MST (PDB: 4JGT) are shown in blue and green, respectively, and the linker is in red. Pyruvate (cyan) and key active site residues including Cys248 in the Cys-SSH state are shown in stick representation. (B) Close up of the active site captured in a product complex with pyruvate (orange) and Cys-SSH (PDB: 4JGT). Two residues in the catalytic triad (D63 and H74) are donated by the N-terminal domain and are shown in blue. (C) Mechanism of the reaction between the 3-mercaptopyruvate substrate and the Cys248 thiolate.
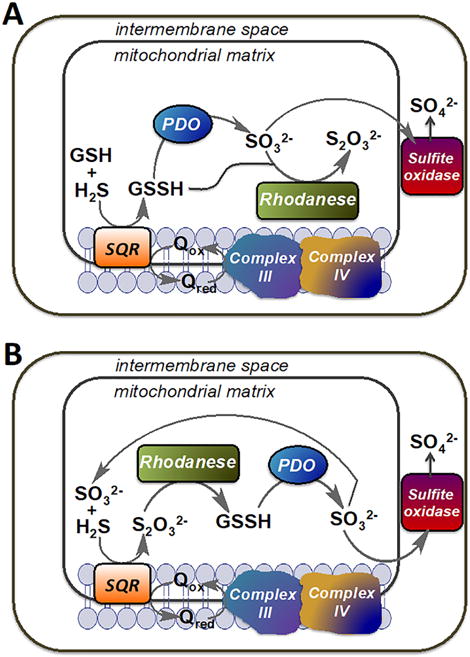
Alternative models describing the organization of the mitochondrial sulfide oxidation pathway. (A) In this model, GSH is the sulfur acceptor from SQR and the product, GSSH, is utilized by either PDO or by rhodanese generating sulfite and thiosulfate, respectively. (B) In this model, sulfite is the sulfur acceptor from SQR and the product, thiosulfate, is utilized by rhodanese to generate GSSH, which is subsequently oxidized by PDO to sulfite. Sulfite is eventually oxidized to sulfate by sulfite oxidase. Q represents coenzyme Q.
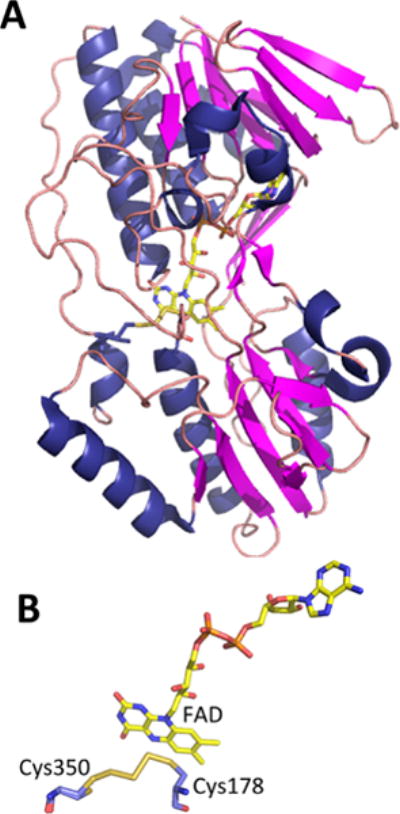
Structure of SQR from A. ambivalens. (A). Structure of an SQR subunit (PDB: 3H8L) in which the covalently bound FAD is shown in stick representation. (B) Close-up of the active site showing FAD and a bridging trisulfide intermediate between Cys350 and Cys178.
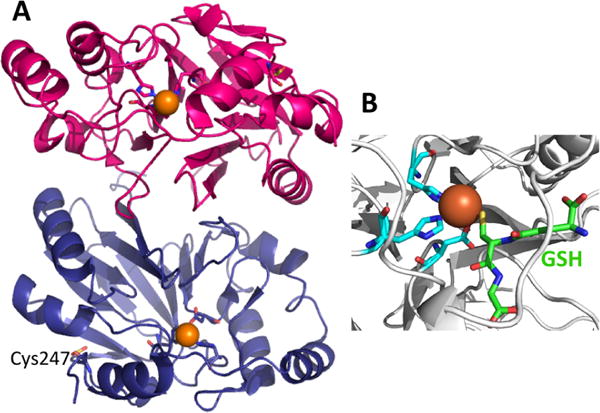
Crystal structure of PDO. (A) Structure of human PDO (PDB: 4CHL) in which the protomers are shown in pink and blue, the iron ion as an orange sphere, and the coordinating histidines and aspartate in stick representation. Cys247 is oxidized as cysteine sulfinate and is also shown in stick representation. (B). Close up of the P. putida PDO (PDB: 4YSL) with bound GSH (green). The cysteine sulfur of GSH is proximal to the iron ion, which is coordinated by a 2His-1Asp facial triad.
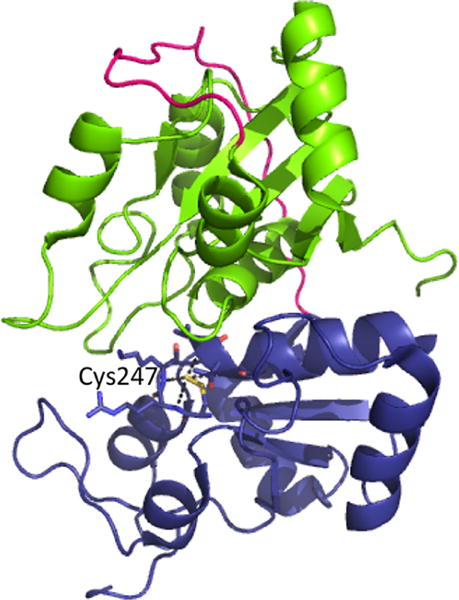
Structure of bovine rhodanese (PDB: 1RHD). The N- and C-terminal domains are shown in green and blue with the intervening linker in pink. A Cys-SSH intermediate is stabilized at Cys247 in the active site, via hydrogen bonding interactions with neighboring residues.
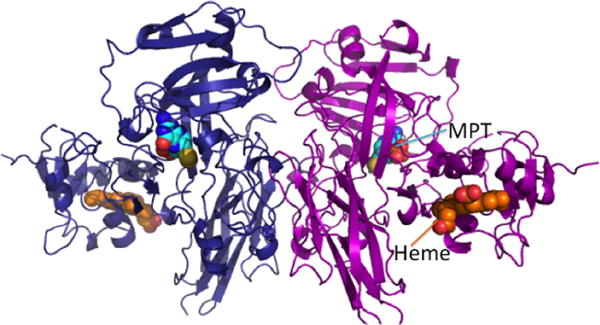
Structure of dimeric chicken sulfite oxidase (PDB: 1SOX). The two subunits are shown in blue and magenta, respectively, and the heme (orange) and molybdopterin (MPT, cyan) cofactors are in sphere representation.
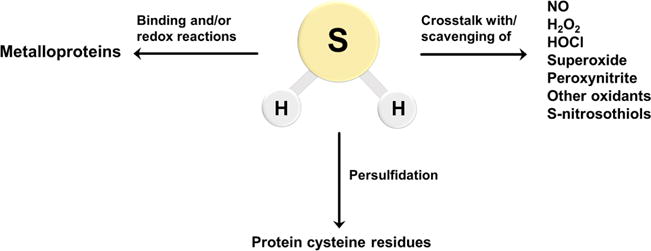
Biological reactivity of H2S. H2S can react directly with oxidants such as superoxide, HOCl, and ONOO−. It can also react with NO• and S-nitrosothiols leading to the formation of other signaling species (right arrow). Metal centers in proteins can bind H2S for delivery to specific targets, be reduced by H2S, or catalyze sulfide oxidation chemistry (left arrow). H2S is also involved in the modification of protein cysteine residues leading to persulfide (Cys-SSH) formation (central arrow).
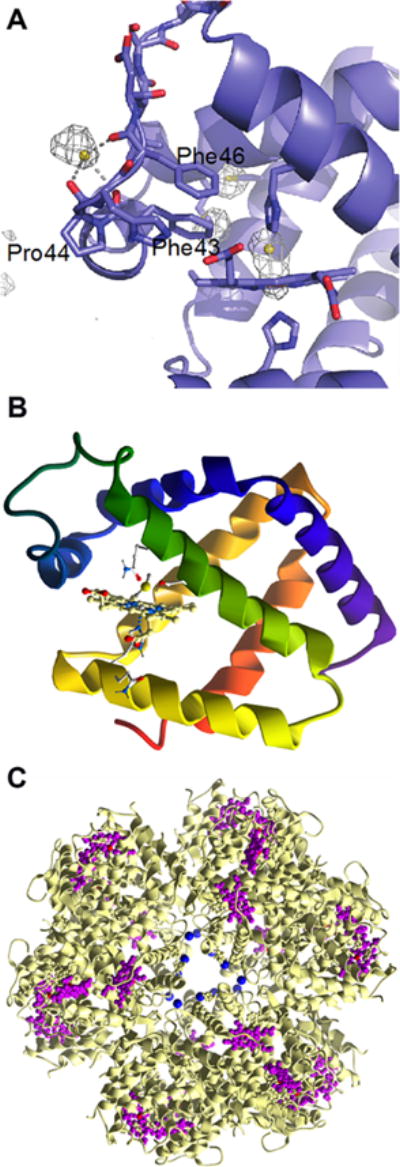
Binding of H2S to protein metal centers. (A) Structure of the α subunit of human hemoglobin showing HS− bound at the entry/exit point of the so-called Phe path that leads to the distal face of the heme. A second sulfide is bound to the heme iron (PDB: 5UCU). (B) Close up of the heme in hemoglobin I from Lucina pectinata with HS− coordinated to the iron ion (PDB: 1MOH). (C) Structure of hemoglobin-like protein C1 from Riftia pachyptila (PDB: 1YHU) with Zn2+ shown in blue and iron hemes in purple.
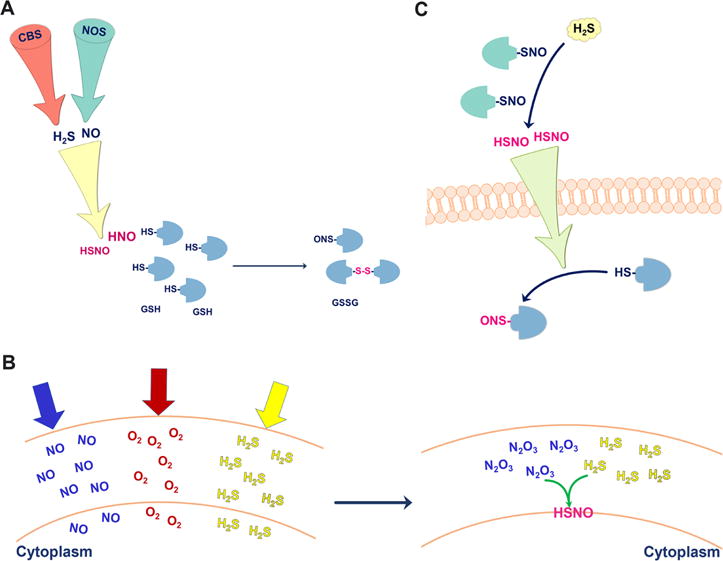
Signaling aspects of NO•/H2S cross-talk. (A) To form HNO and minimize side reactions, H2S and NO2• have to be produced in proximity. HNO (and possibly HSNO) reacts with protein thiols and glutathione. (B) All three gases, NO•, O2, and H2S tend to accumulate in membranes. NO• and O2 form N2O3, which readily reacts with H2S to form HSNO. (C) HSNO formed in the reaction of protein S-nitrosothiols with H2S can diffuse through the cell membrane and transfer the “NO+” group to another protein target.
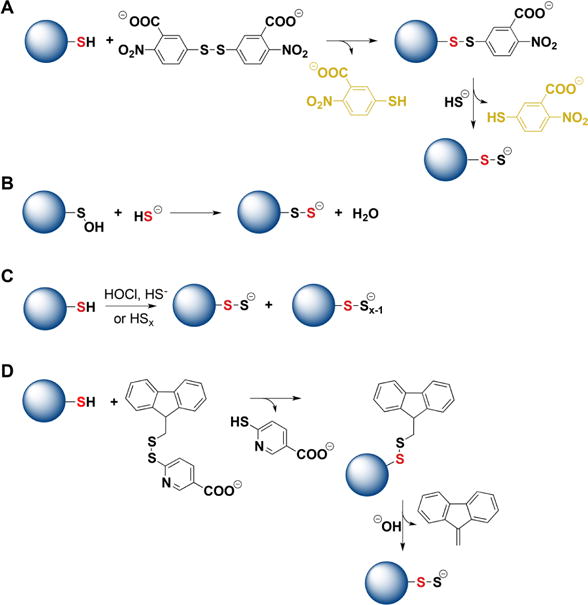
Strategies for the preparation of protein persulfides. (A) Protein thiols react first with DTNB forming mixed disulfides that react with H2S forming persulfides. Thionitrobenzoate is a good leaving group and its UV–visible absorbance can be used to estimate the yield of persulfides. (B) Protein sulfenic acids, when stable, can be used as precursors for persulfide preparation. (C) Protein thiols can be mixed with inorganic polysulfides or with a mixture of HOCl and H2S. Besides persulfides, polythiolated products are also formed. (D) Protein thiols can react with 9-fluorenylmethyl disulfide. The products undergo alkaline hydrolysis forming persulfides.
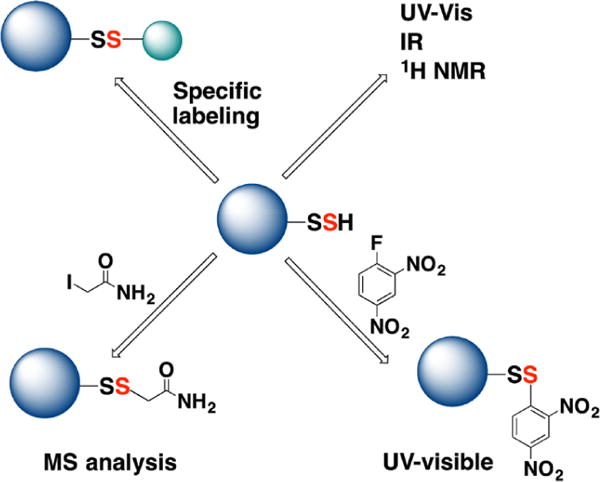
Methodological approaches for the characterization of protein persulfides. (A) When pure, protein persulfides can be characterized by UV–visible, IR, and 1H NMR spectroscopy. (B) Protein persulfides react with 1-fluoro-2,4-dinitrobenzene to form mixed disulfides. DTT releases 2,4-dinitrobenzenethiol, which absorbs at 408 nm under alkaline conditions. (C) Persulfides can be labeled with thiol blocking reagents such as iodoacetamide and analyzed by MS. (D) Protein persulfides can be tagged through different strategies that rely on either their electrophilic or their nucleophilic character.
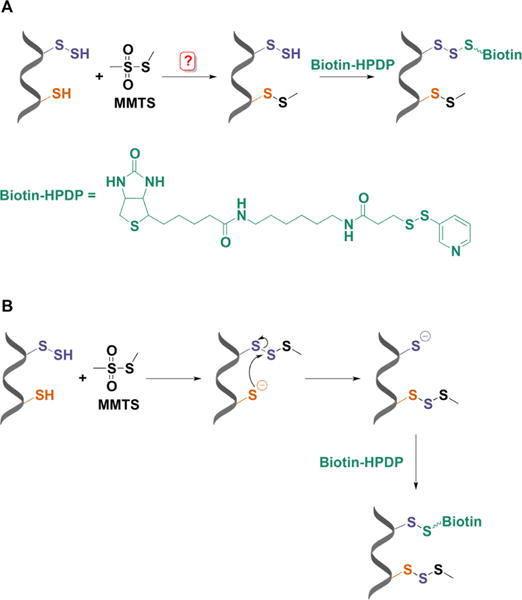
Modified biotin switch assay for persulfide labeling. (A) MMTS was proposed to selectively block free thiols leaving persulfides unmodified and ready for reaction with a biotin-derivatized reactive disulfide (biotin-HPDP). (B) Mechanistic explanation for persulfide labeling with the biotin switch assay. MMTS reacts with persulfides more readily than with thiols forming a trisulfide product. This trisulfide is attacked by unreacted thiols leaving a free cysteine that can react with biotin-HPDP.
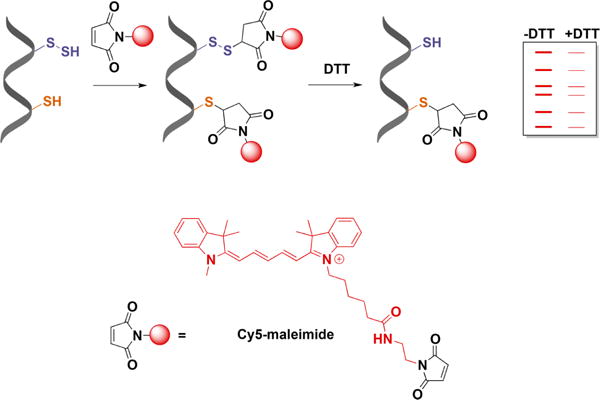
Persulfide detection by differential fluorescence tagging. Both thiols and persulfides are initially blocked with Cy5-maleimide. DTT treatment then removes the fluorescent tag from the persulfides. Proteins are separated by electrophoresis, and the loss of fluorescence caused by DTT is used as a measure of persulfidation.
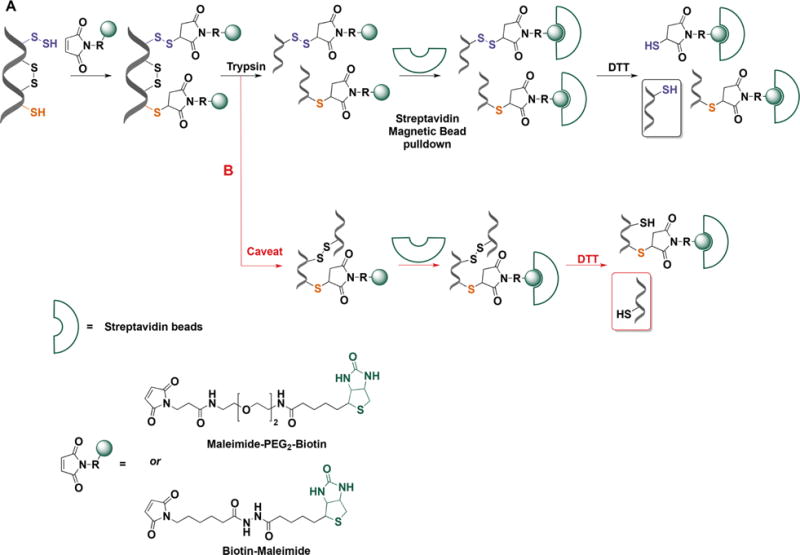
Persulfide labeling with biotin-tagged alkylating reagents. (A) In the first step proteins are mixed with maleimide-biotin (or maleimide-PEG2-biotin) to tag both thiols and persulfides. Proteins are trypsinized and the biotinylated peptides are bound to streptavidin beads. Persulfidated peptides attach to streptavidin beads via disulfide bonds. DTT treatment facilitates elution from the beads and subsequent MS analysis. (B) A possible caveat is that peptides connected by disulfide bonds and containing a thiol or persulfide could be released from the beads with DTT. However, the concentration of disulfide bonds in intracellular proteins is low and this not expected to be a quantitiatvely major drawback of the method.
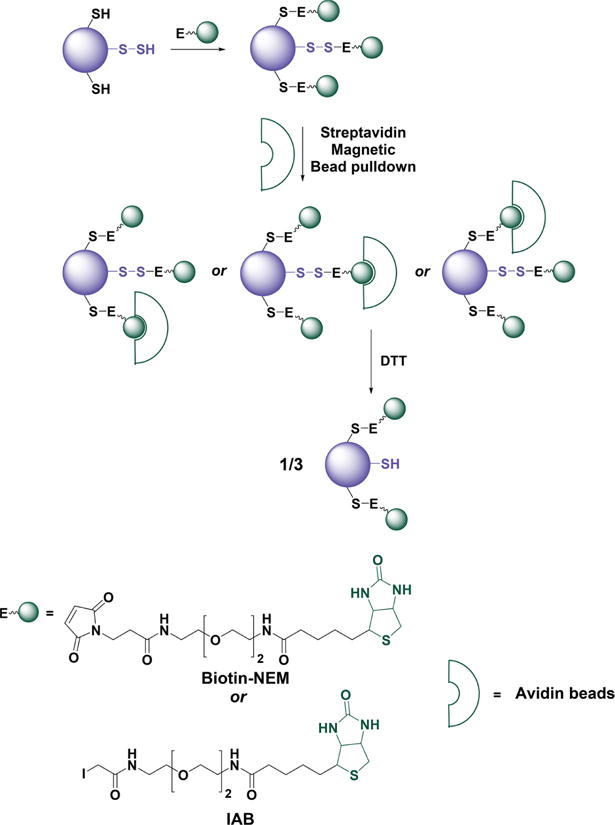
Caveats of whole protein labeling with biotin-tagged alkylating reagents. Proteins containing several cysteine residues, of which only one is persulfidated, are labeled with biotin-maleimide (biotin-NEM) or biotin iodoacetamide (IAB). Since all labels have equal chances of binding to streptavidin beads, the elution with DTT will result in lower than expected yield because the protein will remain bound through the tagged thiols.
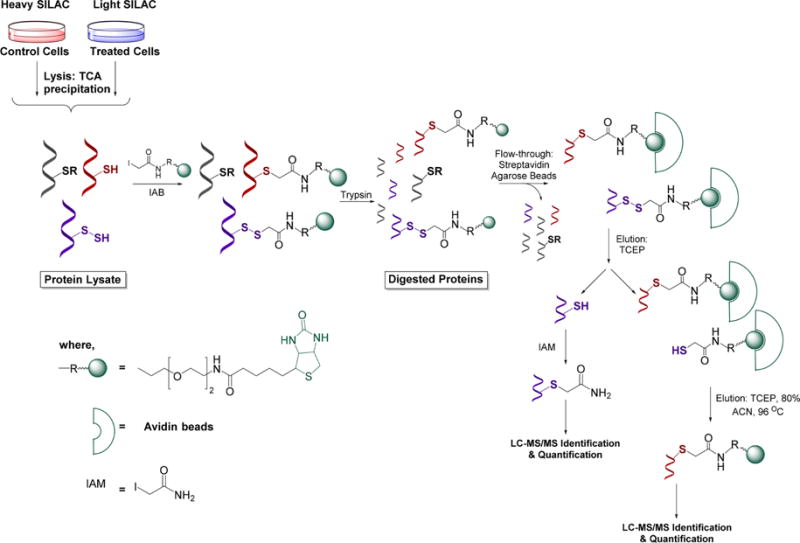
qPerS-SID (quantitative persulfide site identification) approach. Control cells and H2S-treated cells are grown in heavy and light SILAC (stable isotope labeling with amino acids in cell culture) media, respectively. After cell lysis protein extracts are mixed 1:1 and exposed to iodoacetamide-PEG-biotin to labelthiols and persulfides. Proteins are then trypsinized and labeled peptides bound to streptavidinbeads. Since persulfidated peptides attach to streptavidin beads via disulfide bonds they are eluted with TCEP (tris(2-carboxyethyl)phosphine). Released peptides are subjected to LC-MS/MS identification and quantification (enabled by the SILAC approach).
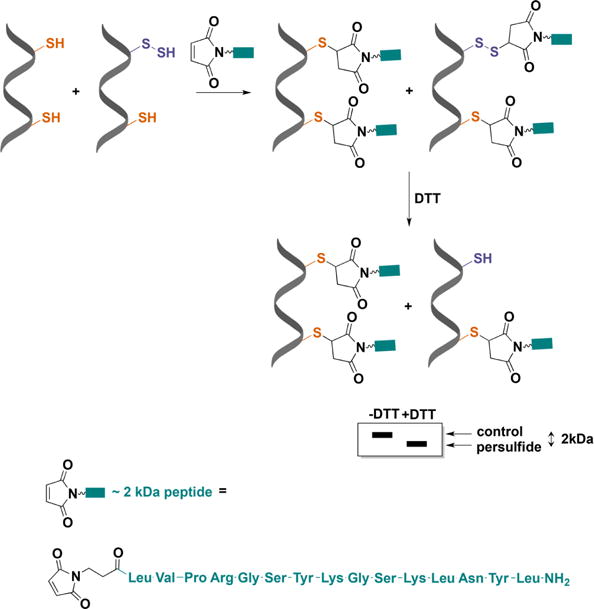
Detection of protein persulfidation by differential peptide tagging. An engineered maleimide with a peptide arm and a molecular mass of ∼2 kDa (MalP) reacts with both thiols and persulfides. DTT removes the mixed disulfides formed with persulfides and increases the electrophoretic mobility of the protein.
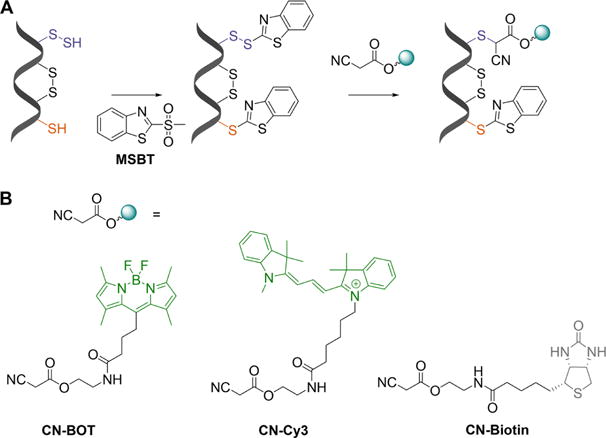
Cyanoacetic acid-based tag-switch method for persulfide labeling. (A) Both persulfides and thiols are initially blocked with MSBT. The product with a persulfidated cysteine is a mixed aromatic disulfide that can be nucleophilically attacked by a cyanoacetic acid–based probe causing a tag-switch. (B) Three different tags are attached to cyanoacetic acid: BODIPY (CN-BOT), Cy3 (CN-Cy3), and biotin (CN-biotin).
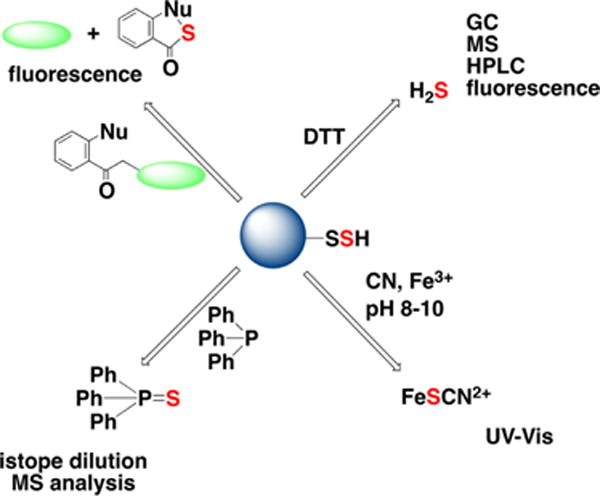
Overview of methodological approaches for total sulfane sulfur detection. Sulfane sulfur compounds release H2S upon DTT treatment. Sulfane sulfur can be detected by the cold cyanolysis method; samples are incubated in alkaline (pH 8–10) cyanide solutions to release SCN−, which is then quantified spectrophotometrically as a complex with Fe3+. Sulfane sulfur can be extracted by triarylphosphines in the form of triarylphosphine sulfide, which can be quantified by isotope dilution MS analysis. Finally, different fluorescence probes (e.g., the SSP series) can detect sulfane sulfur in protein persulfides and low molecultar weight hydropolysulfides in cells (described in greater detail in Chart 28).
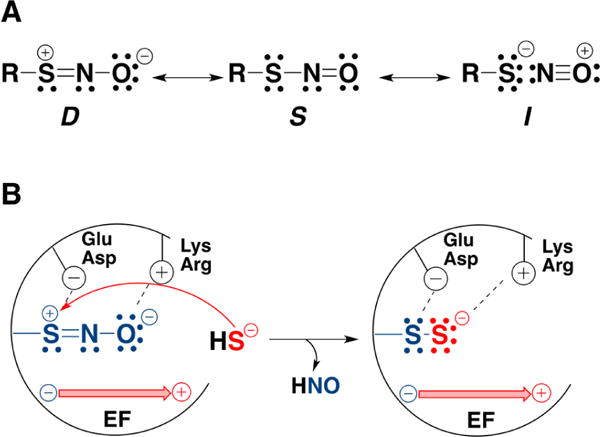
Factors that might favor protein persulfidation over trans-nitrosation in the reaction of an S-nitrosothiol (RSNO) with H2S. (A) Resonance structures of RSNO. Nucleophilic attack on the sulfur is favored in Structure D. (B) Interactions with the protein environment could stabilize the resonance structure D. Positively charged Arg or Lys residues could stabilize the NO moiety, while negatively charged Glu or Asp residues could stabilize the sulfur. In addition electric fields (EF) created by the protein environment could selectively stabilize a resonsance structure, e.g., D, promoting persulfidation and HNO release.
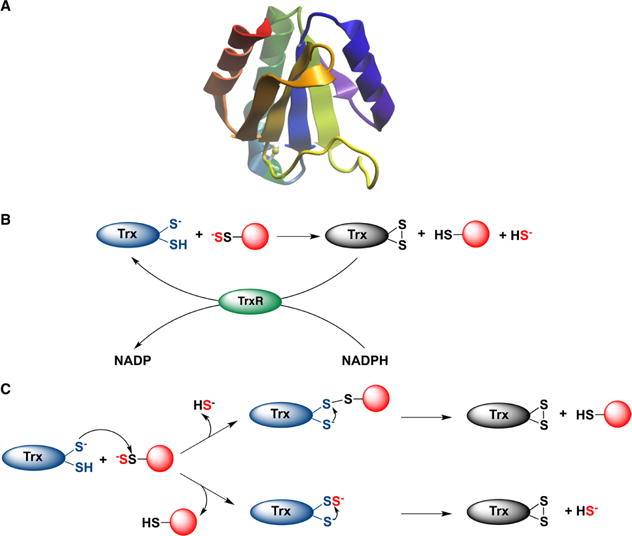
Depersulfidation by thioredoxin (Trx). (A) The structure of human thioredoxin (PDB: 5DQY). (B) Thioredoxin reduces protein persulfides and releases H2S. Oxidized Trx is reduced by thioredoxin reductase (TrxR) at the expense of NADPH. (C) Two possible mechanisms for protein depersulfidation. Top: The nucleophilic thiol attacks the inner sulfur of the protein persulfide forming a mixed protein-Trx disulfide and releasing H2S. In the next step the resolving cysteine reduces the mixed disulfide forming fully oxidized Trx. Bottom: Trx undergoes persulfidation, forming Trx persulfide, which is reduced by the resolving cysteine with concomitant release of H2S.
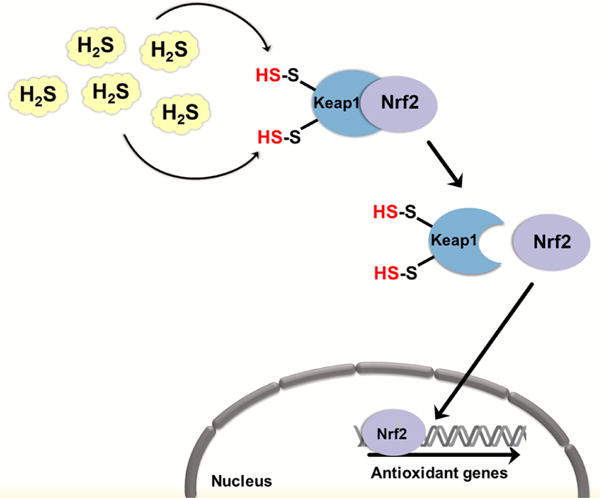
H2S may regulate cellular antioxidant defenses and prevent senescence by persulfidation of Keap1. In the cytosol, Keap1 represses Nrf-2 signaling by binding to it. Bound Nrf-2 is subjected to polyubiquitination and proteasomal degradation. Persulfidation of cysteine residues in Keap1 induces a conformational change, which results in Nrf-2 release. Nrf-2 translocates to the nucleus where it upregulates the expression of various antioxidant defense genes.
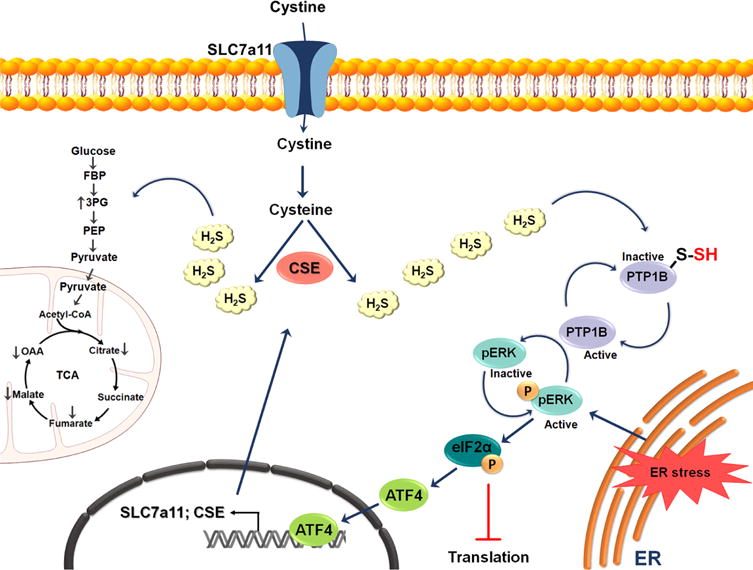
Possible role of H2S in endoplasmic reticulum (ER) stress. Under ER stress, the activity of the transcription factor ATF4 is increased, resulting in the upregulation of CSE and the cystine transporter Slc7a11. The subsequent increased production of H2S leads to the persulfidation of protein tyrosine phosphatase 1B (PTP1B) and consequently to an increase in pERK phosphorylation. pERK activation results in global inhibition of protein translation by activation of eukaryotic translation initiation factor 2α (eIF2α). eIF2α induces ATF4 nuclear translocation. The increased production of H2S during ER stress also results in persulfidation of glycolytic and tricarboxylic acid (TCA) cycle enzymes.
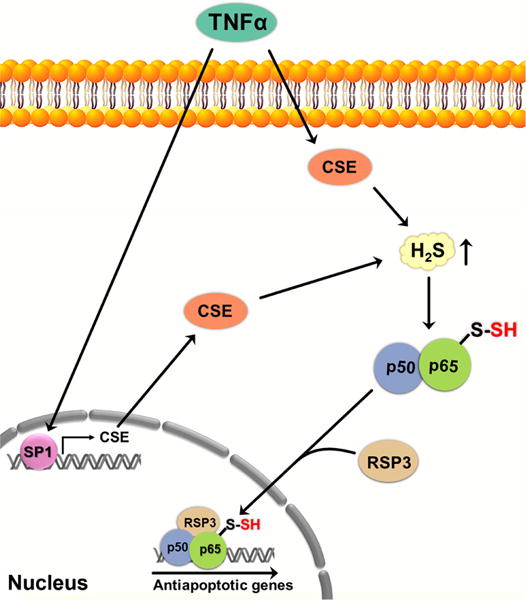
Persulfidation of NF-κB may regulate apoptosis. The proinflammatory cytokine TNFα, involved in the control of inflammatory reactions, stimulates CSE transcription by activating the SP1 transcription factor, resulting in increased H2S levels. H2S induces persulfidation of Cys38 in the p65 subunit of NF-κB, enhancing the binding of NF-κB subunits to the coactivator RPS3. The activator complex then migrates to the nucleus where it upregulates the expression of several antiapoptotic genes. TNFα, tumor necrosis factor α; CSE, cystathionine γ lyase; p50 and p65 subunits of NF-κB; RPS3, ribosomal protein S3; SP1, specificity protein-1.
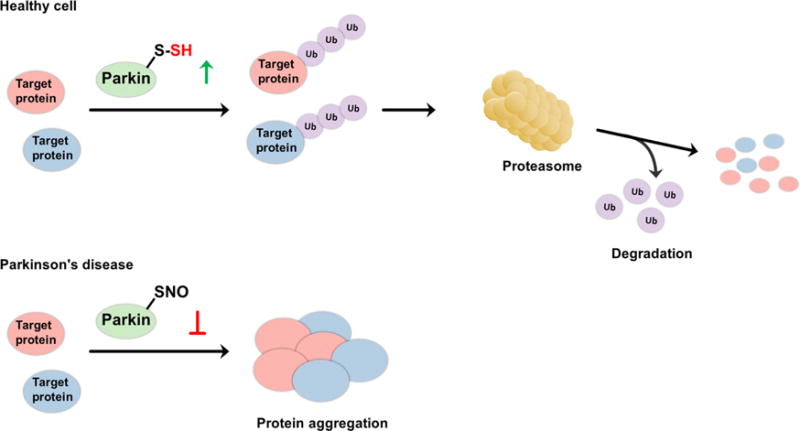
Possible regulatory role of H2S on the catalytic activity of parkin. (A) In healthy subjects, parkin, a E3 ubiquitin ligase, is persulfidated, which increases its enzymatic activity. This leads to ubiquitination of diverse substrates and their subsequent proteasomal degradation. (B) In patients with Parkinson’s disease, parkin is S-nitrosated. The decreased catalytic activity results in protein aggregation, accumulation of toxic proteins, and cell death.
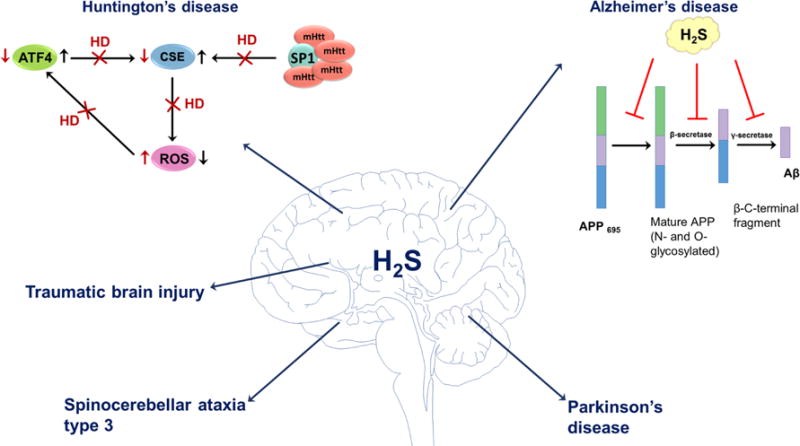
Some possible physiological roles of H2S in neurodegenerative disorders. H2S has been shown to be involved in pathogenesis of Huntington’s, Alzheimer’s, and Parkinson’s diseases, spinocerebellar ataxia, and traumatic brain injury. In healthy subjects, the expression of CSE is regulated by SP1 and ATF4 transcription factors. In Huntington’s disease, abnormal mutated huntingtin (mHtt) protein binds to SP1 and inhibits its activity. Reduced CSE expression results in oxidative stress that subsequently affects ATF4 expression. H2S also inhibits the production of amyloid beta (Aβ) at different catalytic steps of Aβ. The mature isoform of APP is cleaved by β- and γ-secretases forming Aβ. H2S interferes with APP maturation and inhibits the activity of β- and γ-secretases leading to the decreased production of Aβ. In Parkinson’s disease, H2S induces persulfidation of parkin and increases E3 ubiquitin ligase activity. H2S has beneficial effects in spinocerebellar ataxia type 3, where it regulates protein persulfidation and improves SCA3-associated tissue degeneration. In traumatic brain injury H2S exerts antiapoptotic effects and down-regulates the expression of autophagy-related proteins, reduces brain edema and improves the recovery of motor and cognitive dysfunction.
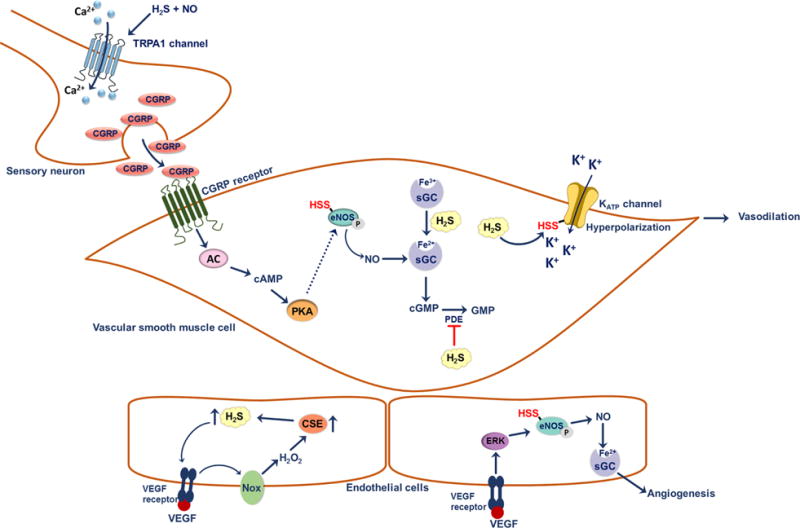
Possible signaling roles of H2S in the vascular system. At a sensory nerve ending, H2S interacts with NO• to give HNO. HNO activates TRPA1 channels; this results in Ca2+ influx and subsequent release of calcitonin gene-related peptide (CGRP). Binding of CGRP to its receptor on vascular smooth muscle cells activates the adenylate cyclase and the cyclic adenosine monophosphate (cAMP)-controlled downstream signaling pathways. As a result of elevated cAMP, protein kinase A (PKA) is activated and could potentially increase the activity of eNOS. Persulfidation of Cys443 on eNOS increases the activity of the enzyme as well as its ability to be phosphorylated, which results in increased production of NO• and activation of soluble guanylate cyclase (sGC). H2S potentiates the binding of NO• to sGC by reducing ferric heme to the ferrous state. Degradation of cGMP by phosphodiesterase (PDE) is prevented by H2S. These effects result in vasodilation of smooth muscle cells. Persulfidation of Cys43 on KATP channel enhances its activity, resulting in the influx of K+ and hyperpolarization of vascular smooth muscle cells. H2S also plays a role in angiogenesis. Binding of VEGF to its receptor on endothelial cells induces the production of H2O2 by activating NADPH oxidase (Nox). H2O2 supposedly increases CSE expression leading in turn, to increased H2S production. H2S stimulates activation of the Akt signaling cascade, which results in the phosphorylation of eNOS, increasing its activity. NO• acts as a pro-angiogenic factor.
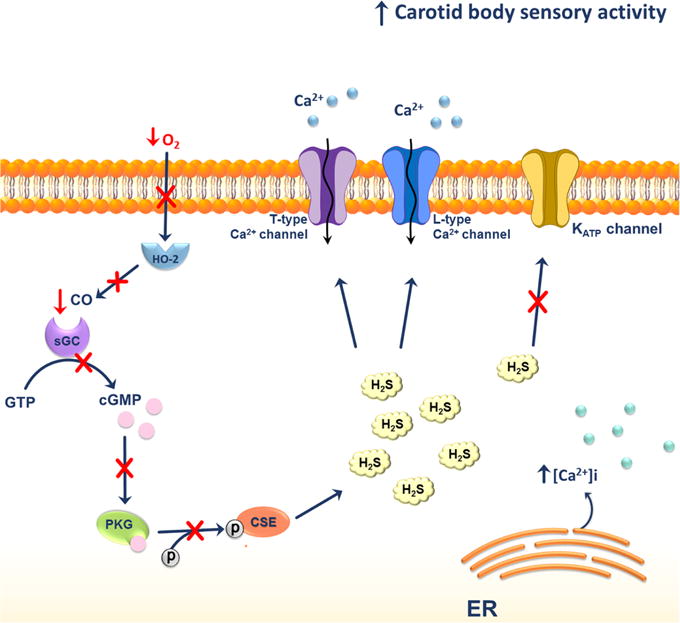
Possible H2S effects on glomus cells of carotid bodies under hypoxic conditions. Under hypoxic conditions the levels of H2S in glomus cells of carotid bodies are elevated. This could be a result of decreased phosphorylation of CSE due to the lack of CO produced by heme oxygenase-2 (HO-2). The lack of CO results in the inhibition of cyclic guanosine monophosphate (cGMP)-stimulated activation of phosphokinase G. The overall increase in H2S levels activates the L-type Ca2+ and T-type voltage-gated Ca2+ channels and mobilizes Ca2+ from the endoplasmic reticulum (ER).

Measurement of H2S through Formation of Methylene Blue
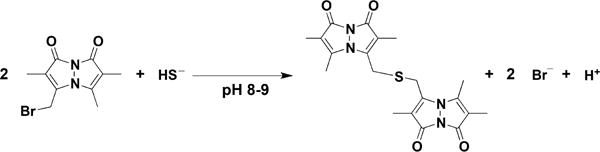
Reaction of Monobromobimane with H2S to form Dibimane Sulfide
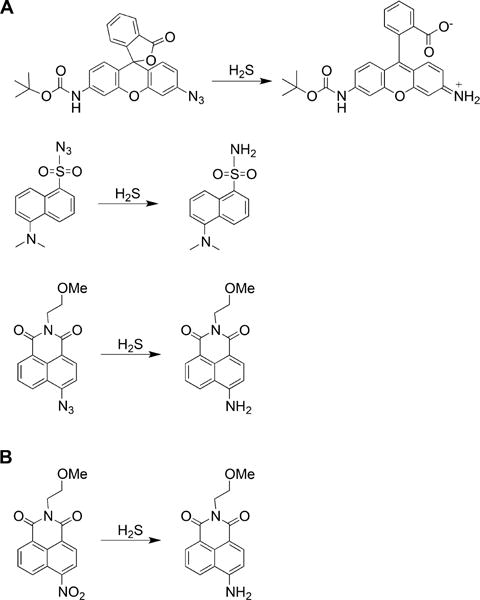
a(A) Reduction of azide groups in rhodamine (top), dansyl (middle), and naphthalimide (bottom) scaffolds. (B) Reduction of nitro group in the naphthalimide scaffold.
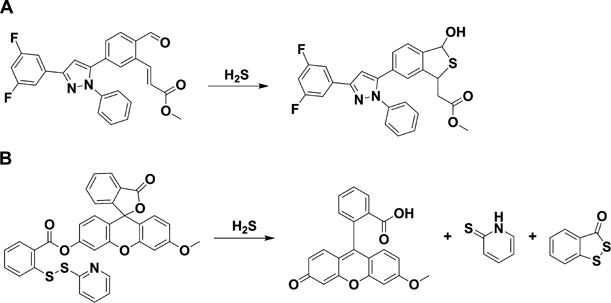
a(A) Probe containing an aldehyde and an acrylate group on a triaryl pyrazoline scaffold. (B) Probe containing an activated disulfide on a fluorescein scaffold.
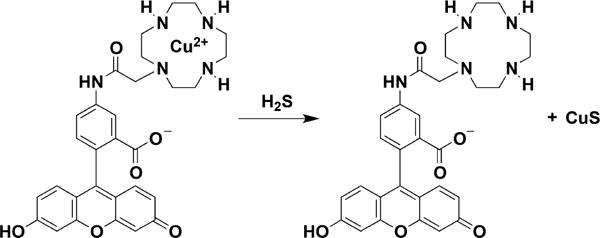
Example of a Probe for H2S Detection Based on the Release of Copper Sulfide from a Cyclen and Fluorescein Derivative
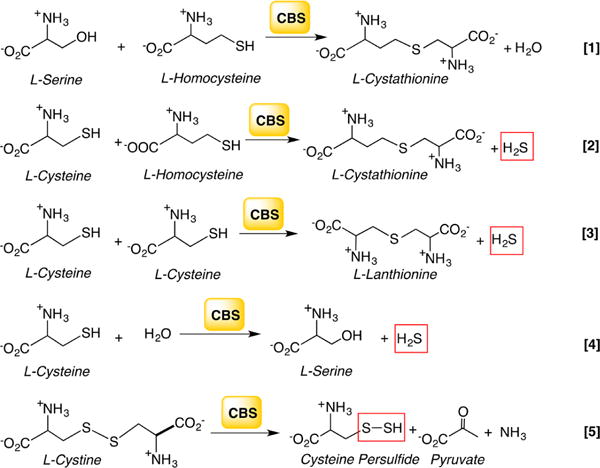
aReaction 1 generates cystathionine in the canonical transsulfuration pathway. Reactions 2–4 generate H2S from cysteine and/or homocysteine, and reaction 5 produces Cys-SSH from cystine.
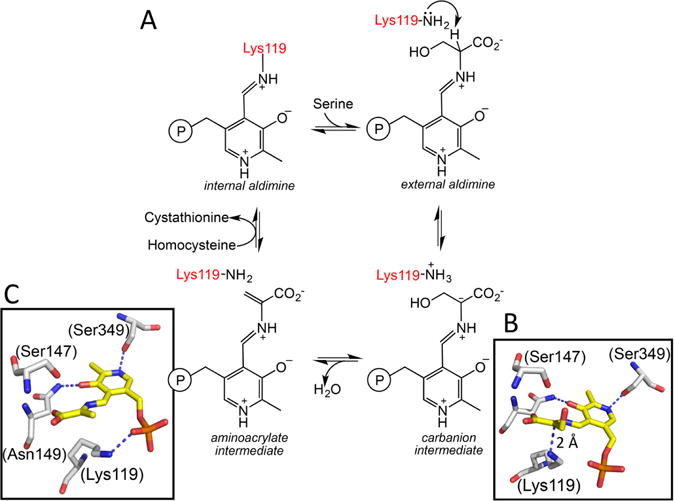
a(A) A minimal mechanism is shown for the β-replacement of serine by homocysteine to generate cystathionine and water. Structures of the carbanion (B) and aminoacrylate (C) intermediates trapped in Drosophila CBS (PDB: 2PC4 and 2PC3) are shown. The numbering of residues shown in parentheses in B and C are for human CBS. The corresponding residues in the fly protein are Lys88, Ser116, Asn118, and Ser318, respectively. An sp2 hybridized α carbon and sp2 hybridized α and β carbons are seen in the carbanion and aminoacrylate intermediates, respectively. Lys119 undergoes a major positional shift in the aminoacrylate intermediate where it is no longer required to stabilize the carbanion.
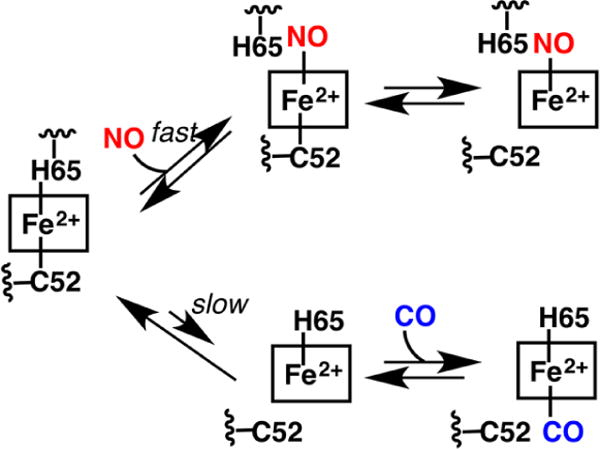
aBinding of NO• is fast and predicted to occur via displacement of the His65 ligand. Binding of CO is slow and limited by the slow dissociation of the Cys52 ligand.
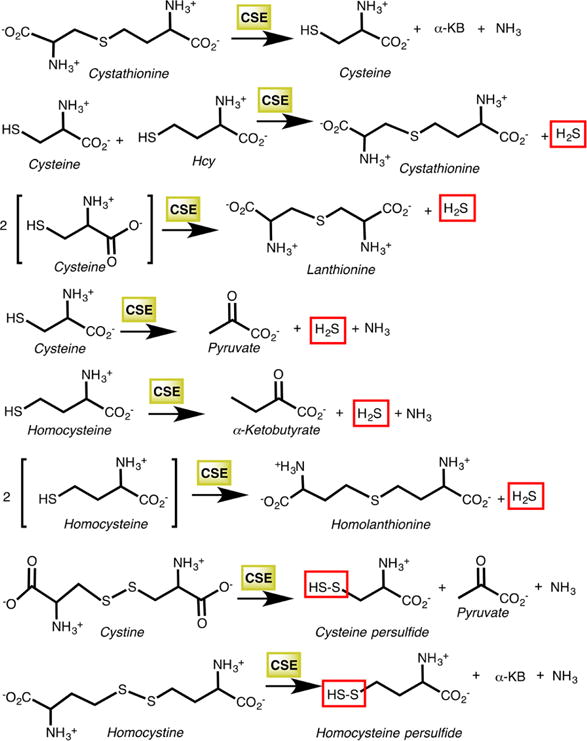
aThe first reaction is the cleavage of cystathionine to cysteine, α-ketobutyrate (α-KB), and ammonia in the canonical transsulfuration pathway. The next five reactions produce H2S, while the last two generate the corresponding persulfides from cystine and homocystine.
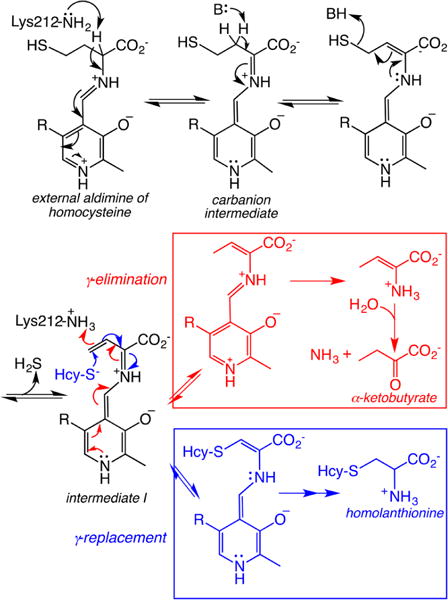
aHcy-S− denotes homocysteine. The first few steps until intermediate I are common to both pathways.
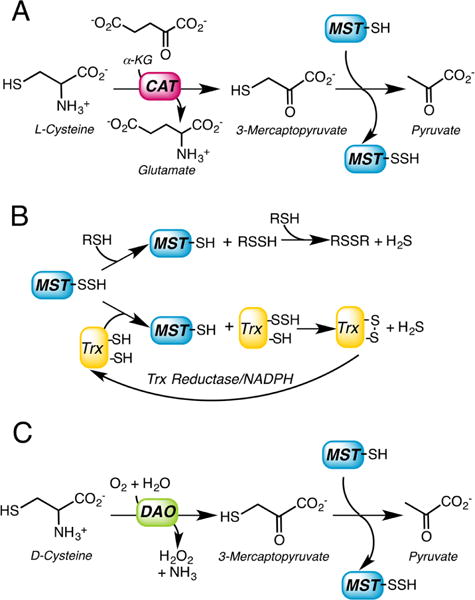
a(A) 3-Mercatopyruvate is synthesized by L-cysteine aminotransferase (CAT), which requires α-ketoglutarate as a cosubstrate. In the first half reaction, MST transfers the sulfur atom from 3-mercaptopyruvate to an active site cysteine forming a Cys-SSH intermediate. (B) In the second half reaction, the outer sulfur from Cys-SSH is transferred to a small molecule thiol acceptor (RSH) or to thioredoxin (Trx) and subsequently released as H2S. (C) 3-Mercatopyruvate can be synthesized from D-cysteine via the action of D-amino acid oxidase (DAO).
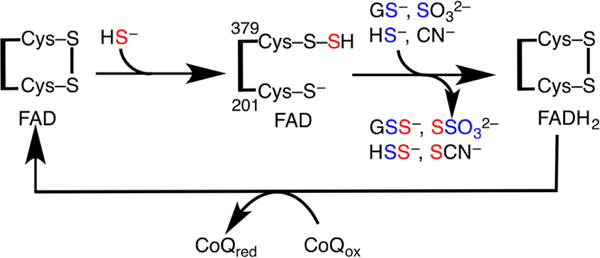
aIn the sulfurtransferase steps, HS− attacks the disulfide bond in SQR forming a persulfide at Cys379 and the sulfane sulfur (in red) is transferred to an acceptor (GSH, sulfite, sulfide, or cyanide). In the electron transfer steps, two electrons are transferred from HS− through the disulfide to FAD and then to CoQ.
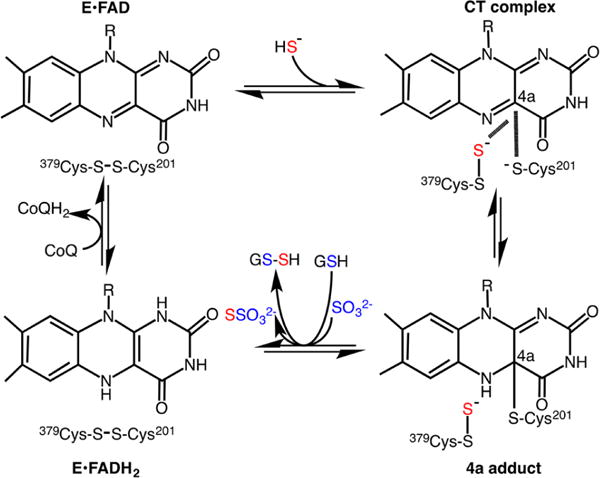
aNucleophilic attack of HS− on the active site disulfide results in the formation of a Cys-SSH intermediate at Cys379 and a charge transfer (CT) complex, which collapses to a postulated 4a adduct. A sulfur acceptor viz. GSH or sulfite moves the sulfane sulfur from the active site, forming GSSH or thiosulfate (S2O32−), respectively, and restoring the active site disulfide. Electron transfer from the reduced flavin, FADH2, to CoQ completes the catalytic cycle.
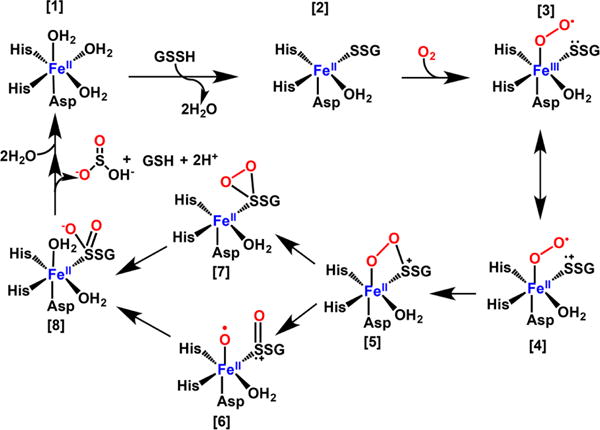
aBinding of GSSH to the resting enzyme [1] creates a binding site for O2 [2]. Formation of a superoxo-FeIII [3] in resonance with a biradical FeII species [4] leads to formation of a cyclic peroxo-FeII species [5]. Cleavage of the O–O bond gives [6]. Alternatively, cleavage of the Fe–O bond gives [7]. Binding of H2O [8], sets up hydrolysis and formation of the product, sulfite. Alternatively, the water that remained coordinated to the metal center could be used for the final hydrolysis step.
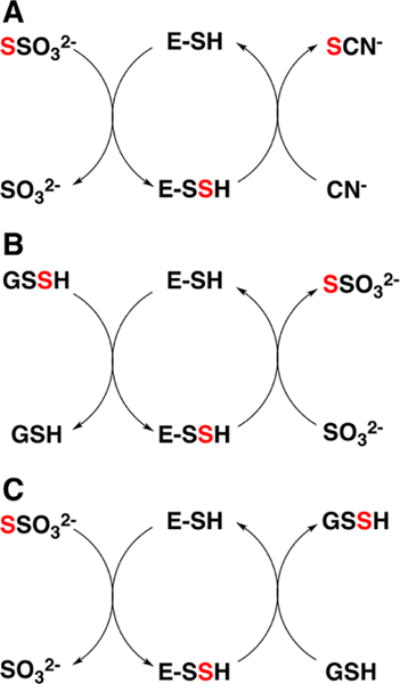
aRhodanese exhibits varied sulfur transferase activities including: (A) thiosulfate:cyanide sulfurtransferase, (B) GSSH:sulfite sulfurtransferase, and (C) thiosulfate:GSH sulfurtransferase. E-SSH denotes the enzyme-bound Cys-SSH intermediate. The red color traces the fate of the sulfur from the donor to the acceptor.
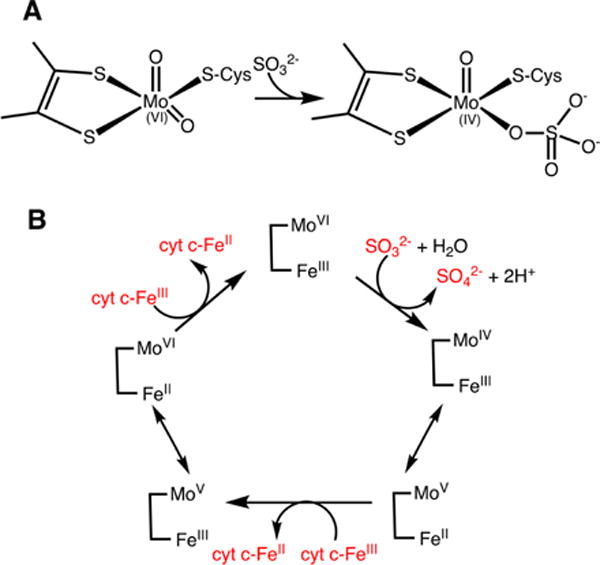
a(A) Attack of sulfite on an oxo/hydroxyl ligand reduces the molybdenum ion. (B) The reaction cycle of sulfite oxidase involves an initial two-electron reduction of the molybdenum center, which is subsequently oxidized in two one-electron steps via intramolecular electron transfer to the heme. The latter in turn, transfers electrons to the heme in cytochrome c (cyt c) in an intermolecular process.
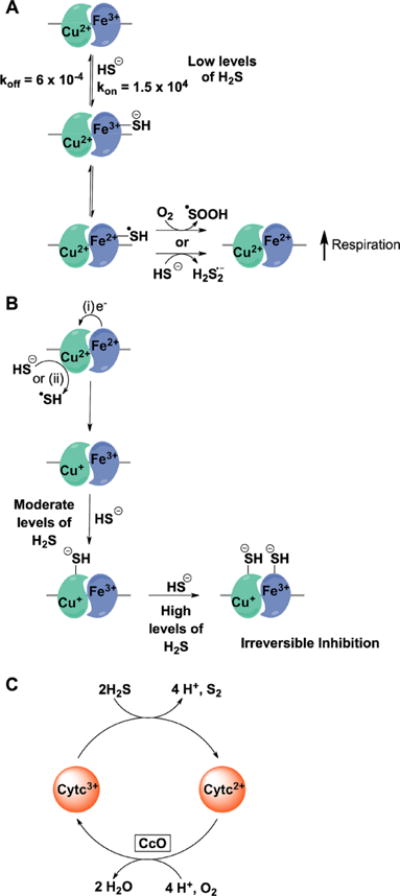
a(A) At low concentrations, H2S binds to ferric heme a3 and reduces it with concomitant formation of HS• which can react with either another molecule of H2S or with oxygen. Reduction of heme a3 leads to an increase in oxygen consumption. (B) At moderate levels, H2S interacts with the CuB+ center forming a stable CuB–SH− complex, which is difficult to oxidize. CuB+ is formed by electron transfer from ferrous heme a3 or by a direct reduction by H2S. At higher levels of H2S, the CuB–SH− complex induces a conformational change and causes further binding of H2S to ferric heme a3. (C) Alternatively, H2S can reduce cytochrome c thereby increasing CcO reduction and respiration.
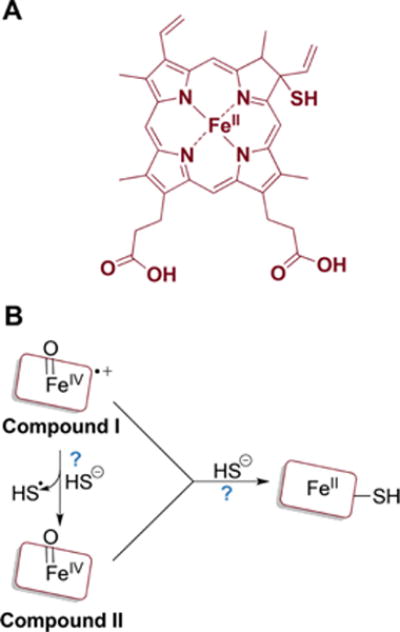
a(A) One of the proposed structures of sulfheme. (B) The mechanism of sulfheme formation is not fully understood (as denoted by the question marks) but starts with compound I or II reacting with H2S and results in sulfur being incorporated into the porphyrin ring.
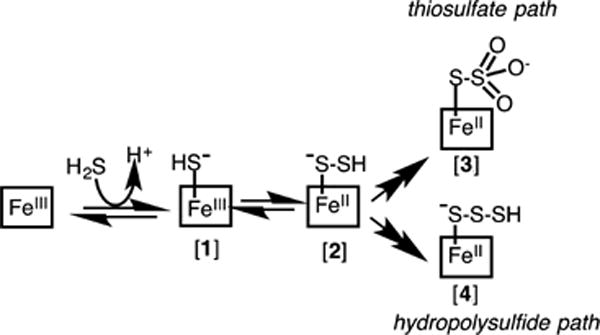
aDetails of the oxidation chemistry that lead to the products have been omitted for clarity.
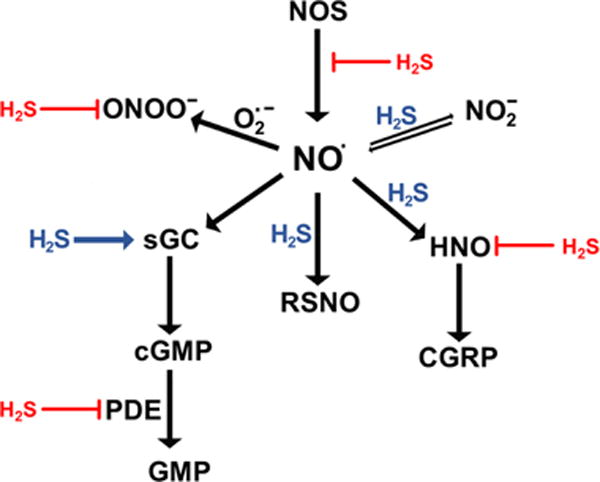
aNO• signals via the classical soluble guanylate cyclase (sGC)/cyclic GMP (cGMP) cascade. H2S can reduce sGC to increase NO• binding and cGMP production. cGMP is deactivated by phosphodiesterase 5 (PDE), an enzyme that is inhibited by H2S. Oxidation of NO• to nitrosonium (NO+) ion leads to modification of cysteine residues and formation of S-nitrosothiols (RSNO), a process that H2S can facilitate. NO• can be reduced by H2S to form HNO. HNO activates the release of calcitonin gene-related peptide (CGRP), a vasodilator, but HNO can also be trapped by H2S. NO• is oxidized to nitrite, which can be reduced back to NO•, a process that H2S can facilitate. NO• reacts with superoxide to form peroxynitrite (ONOO−). H2S can scavenge ONOO−.
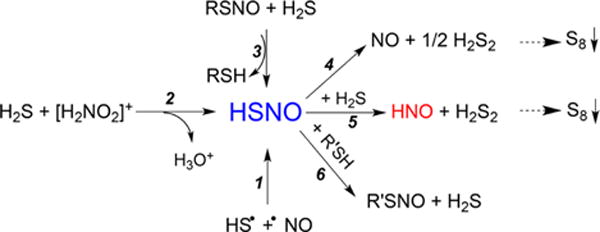
Reaction Paths for HSNO Generation (1–3) and Its Biologically Relevant Reactions (4–6)
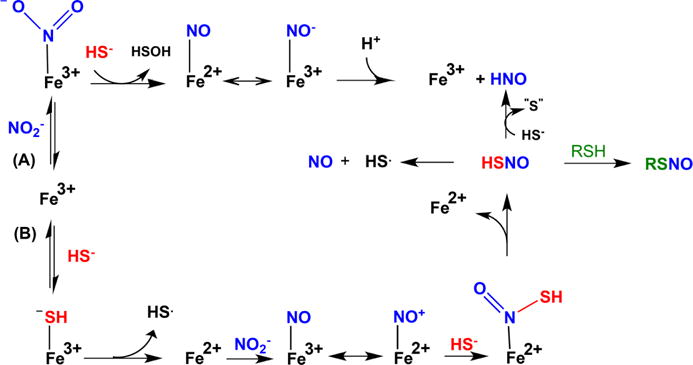
aPathway A, which predominates when nitrite is in excess over H2S, represents a classical oxygen atom transfer; nitrite coordination to ferric heme leads to HSOH and [Fe2+(NO)] ↔ [Fe3+(NO−)], which releases HNO slowly. Pathway B predominates when H2S is in excess over nitrite; reduction of ferric heme by H2S is followed by nitrite reduction to [Fe3+(NO)] ↔ [Fe2+(NO+)] species, which is scavenged by HS− giving HSNO. Either free or coordinated HSNO causes transnitrosation of protein thiols or, in the reaction with H2S, generates HNO.
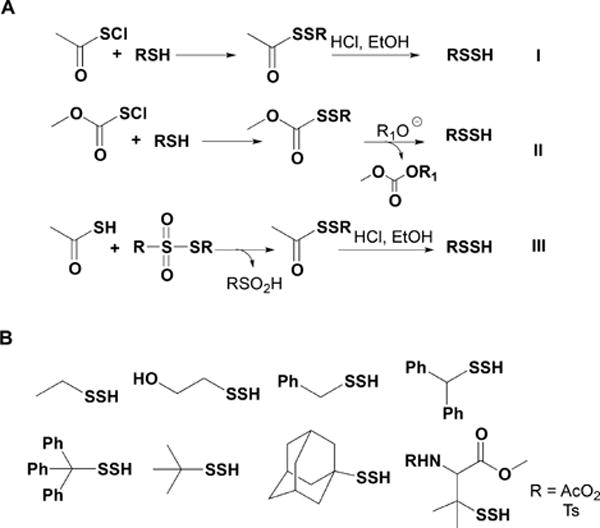
Synthetic Strategies for the Preparation of Low Molecular Weight Persulfides (A) and Structures of Some of the Persulfides Prepared following These Synthetic Routes (B)
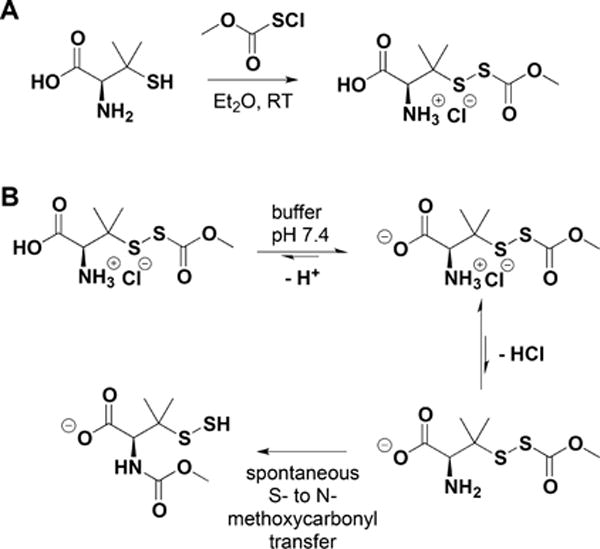
Synthesis of an Acyl-Protected Disulfide of Penicillamine (A) and Its Rearrangement to N-Methoxycarbonyl Penicillamine Persulfide via S- to N-Methoxycarbonyl Transfer at pH 7.4 (B)
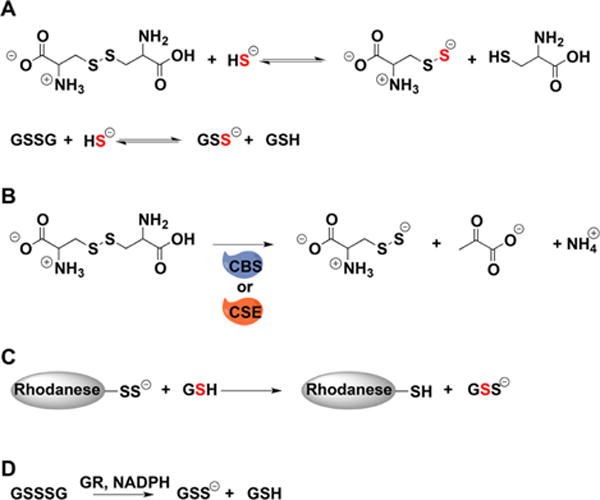
a(A) Cysteine and glutathione persulfides can be prepared by the reaction of H2S with cystine and glutathione disulfide, respectively. (B) Cysteine persulfide can be generated from cystine and CSE or CBS. (C) Rhodanese can be used to transfer sulfur to glutathione. (D) Glutathione reductase (GR) uses electrons from NADPH to reduce glutathione trisulfide to persulfide and GSH.
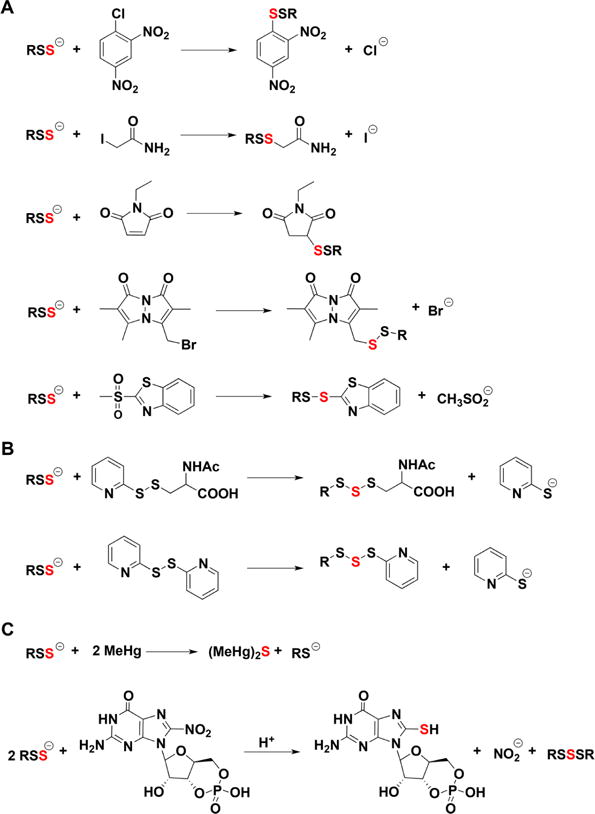
a(A) Reactions with thiol alkylating agents. (B) Reactions with disulfides. (C) Reactions with methylmercury and nitroguanosine 3′,5′-cyclic monophosphate.
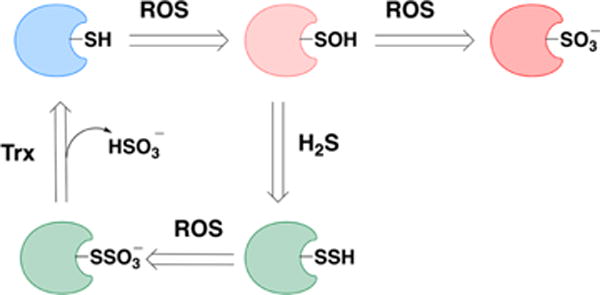
aA thiol can be oxidized to a sulfenic acid. The latter can be reduced back to thiol or be further oxidized to sulfonate, an irreversible modification. Persulfides, if exposed further to oxidants, will form S-sulfocysteines (−SSO3−). Enzymes such as thioredoxin can reduce the S–S bonds and restore the native thiol.
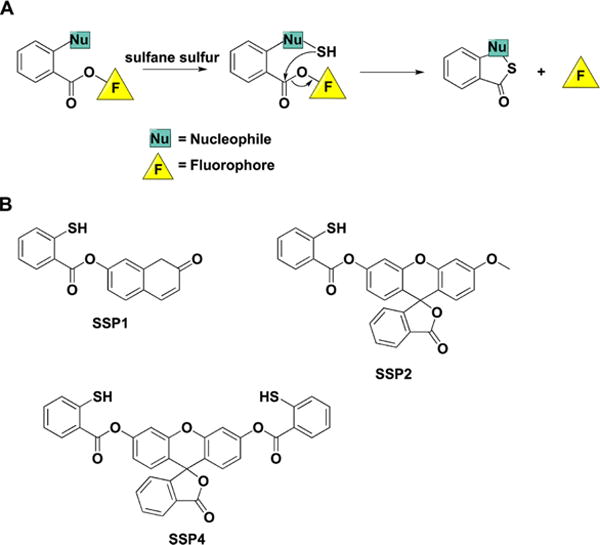
a(A) Internal cyclization and fluorophore release following the initial reaction of the probe with sulfane sulfur compounds. (B) Structures of the synthetic probes that exhibit this type of chemical reactivity.
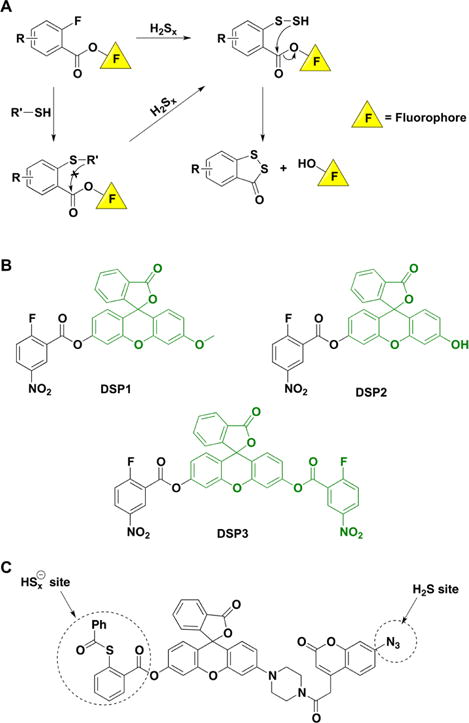
a(A) Reactions leading to fluorophore release. (B) Structures of the synthetic probes that exhibit this type of reactivity. (C) FRET-based probe for the simultaneous detection of H2S and sulfane sulfur-containing species.
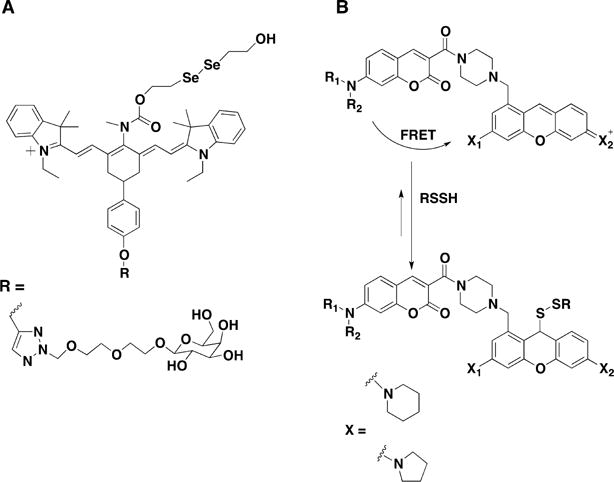
Ratiometric Near-IR Fluorescence Probe for Cysteine Persulfide Detection (A) and FRET Probe Designed for Persulfide Detection (B)
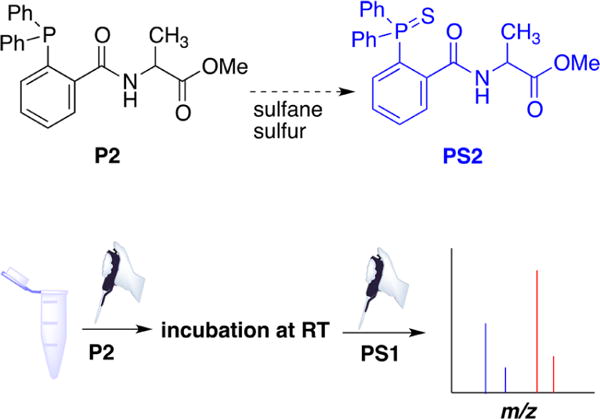
aThe reaction of triarylphosphine (P2) with sulfane sulfur compounds. After incubation of P2 with cell or tissue samples to form PS2, PS1 (13C-labeled triarylphosphine sulfide) is added as internal standard and the samples analyzed by MS.
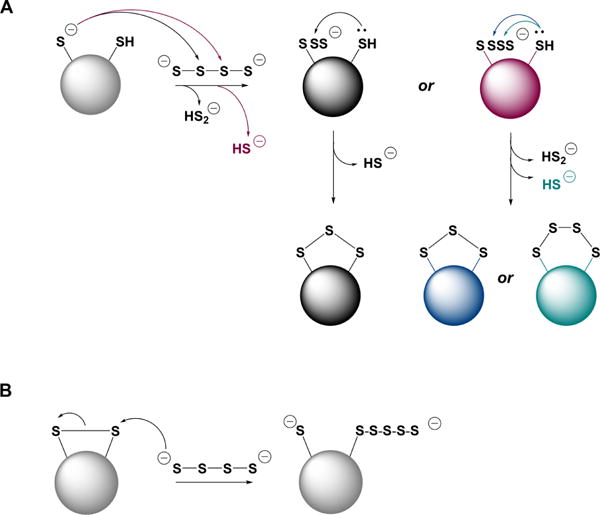
a(A) Inorganic polysulfides can react as electrophiles with a protein thiolate. The resulting polythiolated cysteine can have different numbers of S atoms and can in turn, be attacked by a proximal cysteine forming a family of products, e.g., intramolecular disulfides, trisulfides, etc. (B) Inorganic polysulfides can react as nucleophiles with an intramolecular protein disulfide forming thiol and polythiolated cysteine.
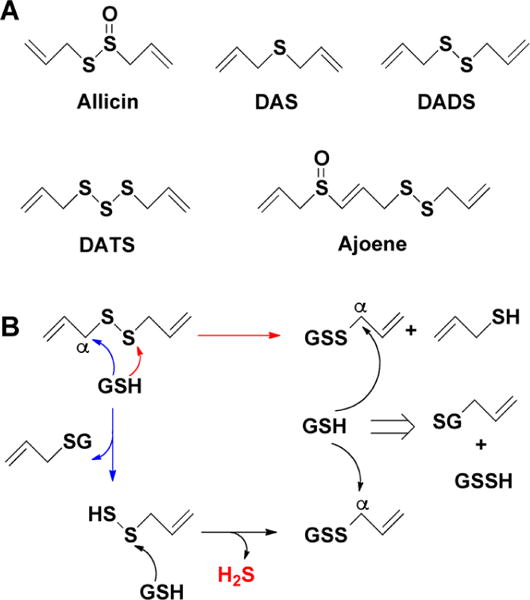
a(A) Allicin (diallyl thiosulfinate) is rapidly metabolized in aqueous solutions into diallyl sulfide (DAS), diallyl disulfide (DADS), diallyl trisulfide (DATS), and ajoene. (B) Glutathione-promoted decomposition of DADS and generation of allylpersulfide, glutathione persulfide (GSSH), and H2S.
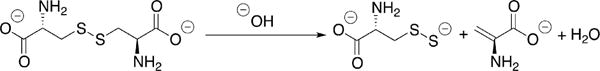
Base-Promoted Persulfide Generation from Cystine
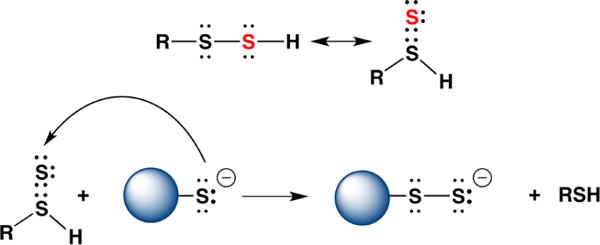
Tautomerization of a Persulfide to a Thiosulfoxide as a Postulated Mechanism for Transpersulfidation
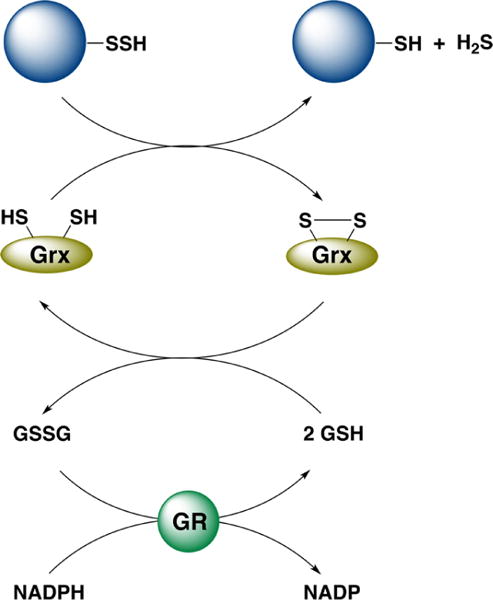
aGlutaredoxin (Grx) reduces a protein persulfide, oxidized Grx is reduced by glutathione (GSH), and GSSG is reduced by glutathione reductase (GR) at the expense of NADPH.
References
-
- Winogradsky S. Ueber Schwefelbakterien. Bot Zeitung. 1887;45:489–507. 513–523, 529–539, 545–559, 569–576, 585–594.
-
- Ramazzini B. Baptiftam Conzattum; Padua: 1713. De Morbis Artificum Diatriba.
-
- Scheele CW. Chemische Abhandlung von der Luft und dem Feuer (Chemical treatise on air and fire) Magnus Swederus; Upsala, Sweden: 1777.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
