

Solution structure of an ultra-stable single-chain insulin analog connects protein dynamics to a novel mechanism of receptor binding
- PMID: 29114034
- PMCID: PMC5766920
- DOI: 10.1074/jbc.M117.808667
Solution structure of an ultra-stable single-chain insulin analog connects protein dynamics to a novel mechanism of receptor binding
Abstract
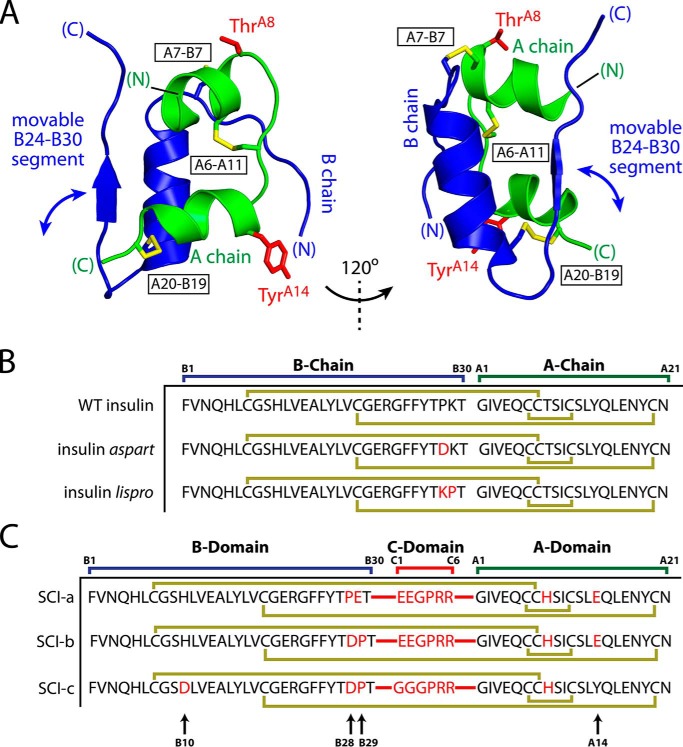
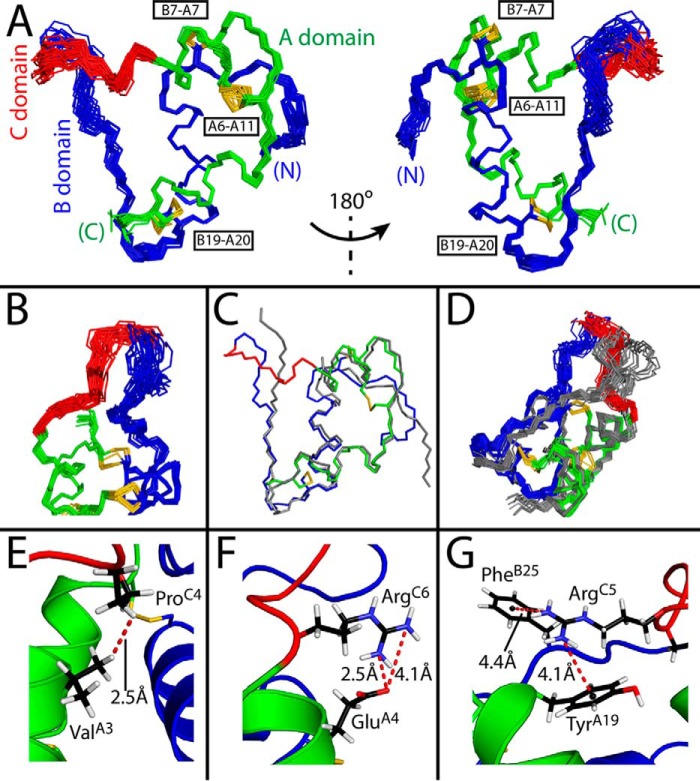
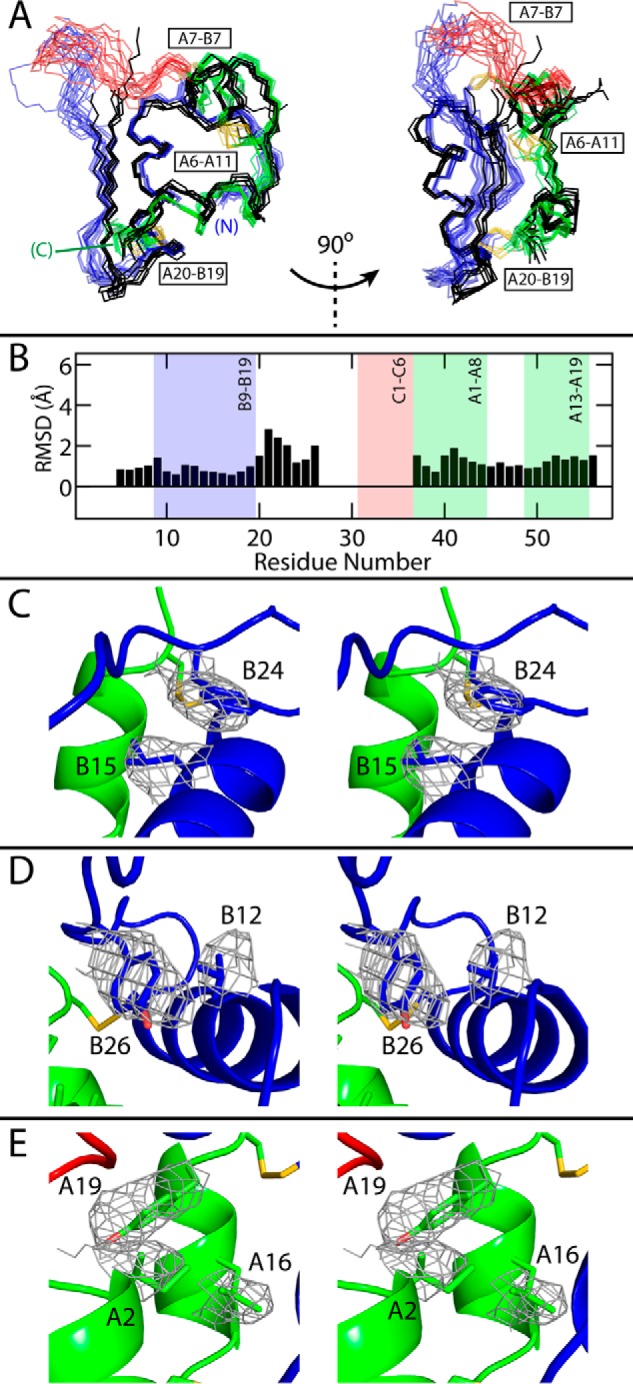
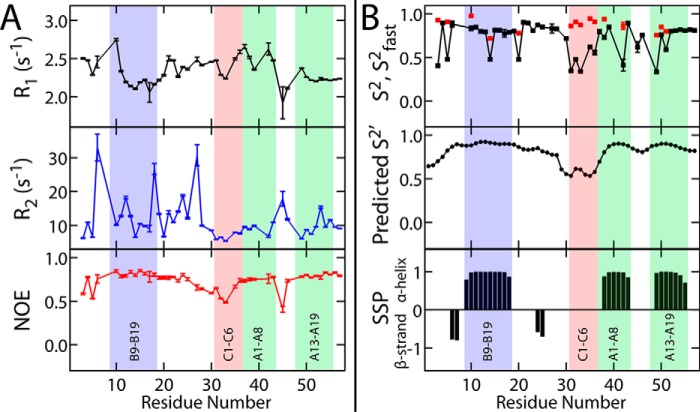
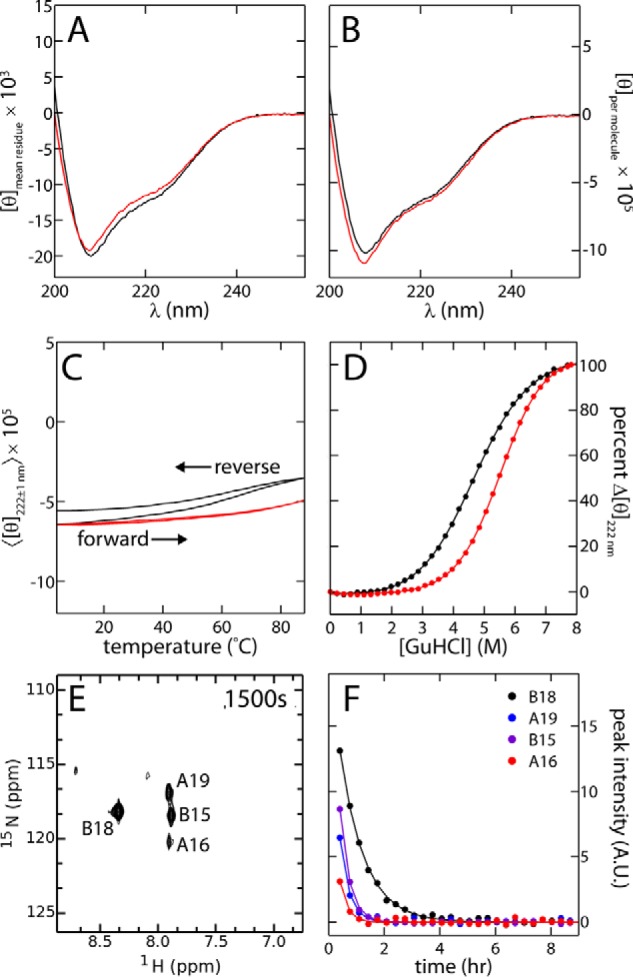
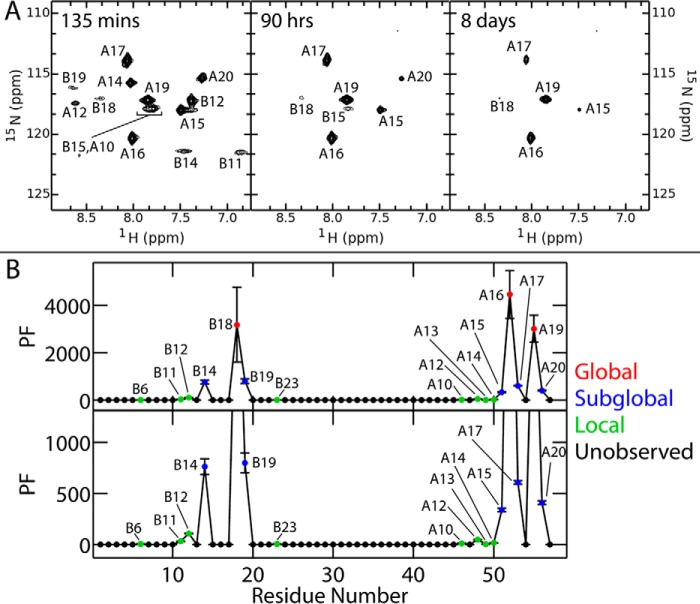
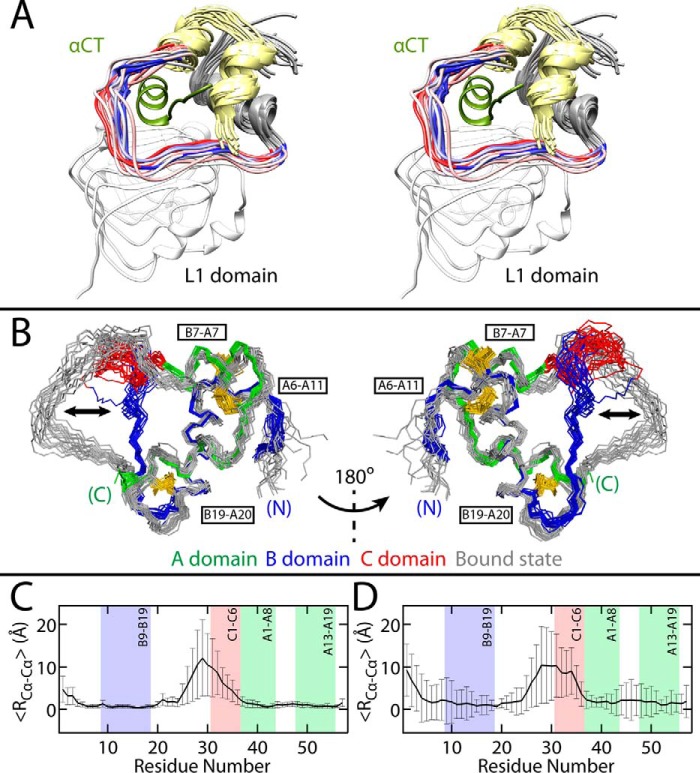
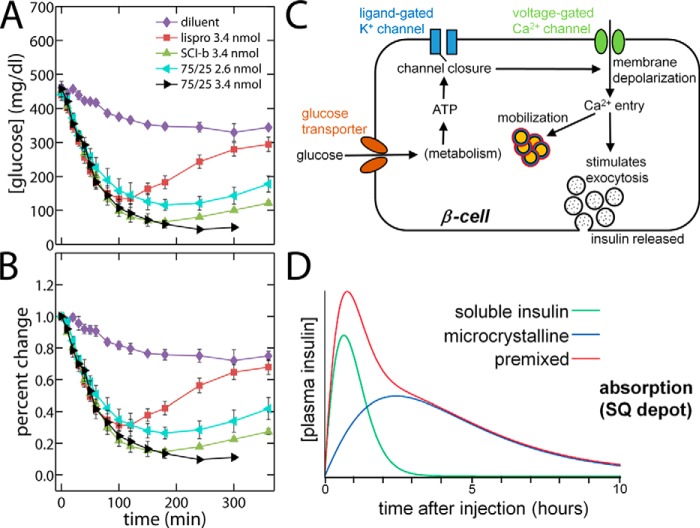
Domain-minimized insulin receptors (IRs) have enabled crystallographic analysis of insulin-bound "micro-receptors." In such structures, the C-terminal segment of the insulin B chain inserts between conserved IR domains, unmasking an invariant receptor-binding surface that spans both insulin A and B chains. This "open" conformation not only rationalizes the inactivity of single-chain insulin (SCI) analogs (in which the A and B chains are directly linked), but also suggests that connecting (C) domains of sufficient length will bind the IR. Here, we report the high-resolution solution structure and dynamics of such an active SCI. The hormone's closed-to-open transition is foreshadowed by segmental flexibility in the native state as probed by heteronuclear NMR spectroscopy and multiple conformer simulations of crystallographic protomers as described in the companion article. We propose a model of the SCI's IR-bound state based on molecular-dynamics simulations of a micro-receptor complex. In this model, a loop defined by the SCI's B and C domains encircles the C-terminal segment of the IR α-subunit. This binding mode predicts a conformational transition between an ultra-stable closed state (in the free hormone) and an active open state (on receptor binding). Optimization of this switch within an ultra-stable SCI promises to circumvent insulin's complex global cold chain. The analog's biphasic activity, which serendipitously resembles current premixed formulations of soluble insulin and microcrystalline suspension, may be of particular utility in the developing world.
Keywords: diabetes; hormone; protein engineering; protein structure; receptor tyrosine kinase.
© 2018 by The American Society for Biochemistry and Molecular Biology, Inc.
Conflict of interest statement
M. A. W. has equity in Thermalin Diabetes, LLC (Cleveland, OH), where he serves as Chief Innovation Officer; he has also been a consultant to Merck Research Laboratories and DEKA Research & Development Corp. N. B. P. and F. I.-B. are consultants to Thermalin Diabetes, LLC. Part of M.C.L.'s research is funded by Sanofi (Germany)
Figures
References
-
- Brange J., Ribel U., Hansen J. F., Dodson G., Hansen M. T., Havelund S., Melberg S. G., Norris F., Norris K., and Snel L. (1988) Monomeric insulins obtained by protein engineering and their medical implications. Nature 333, 679–682 - PubMed
-
- Weiss M. A. (2013) Design of ultra-stable insulin analogues for the developing world. J. Health Spec. 1, 59–70 10.4103/1658-600X.114683 - DOI
-
- Vinther T. N., Norrman M., Ribel U., Huus K., Schlein M., Steensgaard D. B., Pedersen T. Å., Pettersson I., Ludvigsen S., Kjeldsen T., Jensen K. J., and Hubálek F. (2013) Insulin analog with additional disulfide bond has increased stability and preserved activity. Protein Sci. 22, 296–305 10.1002/pro.2211 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
