

The JNK Signaling Pathway in Renal Fibrosis
- PMID: 29114233
- PMCID: PMC5660697
- DOI: 10.3389/fphys.2017.00829
The JNK Signaling Pathway in Renal Fibrosis
Abstract
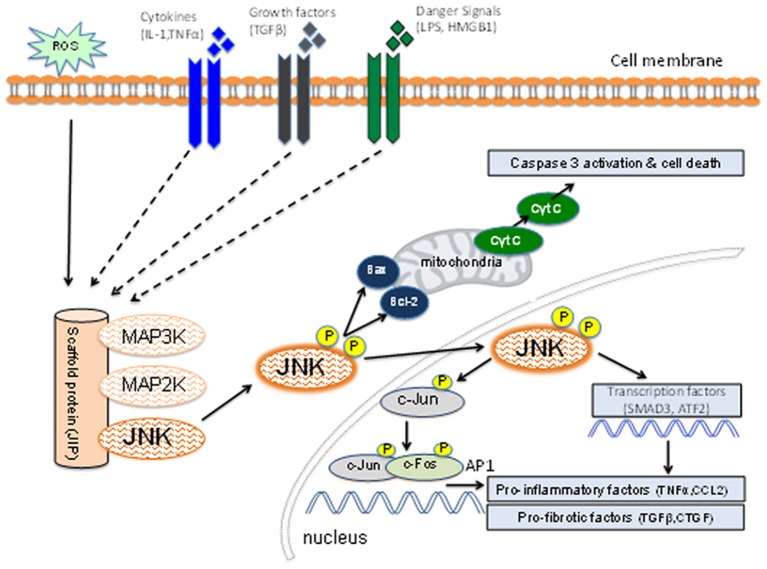
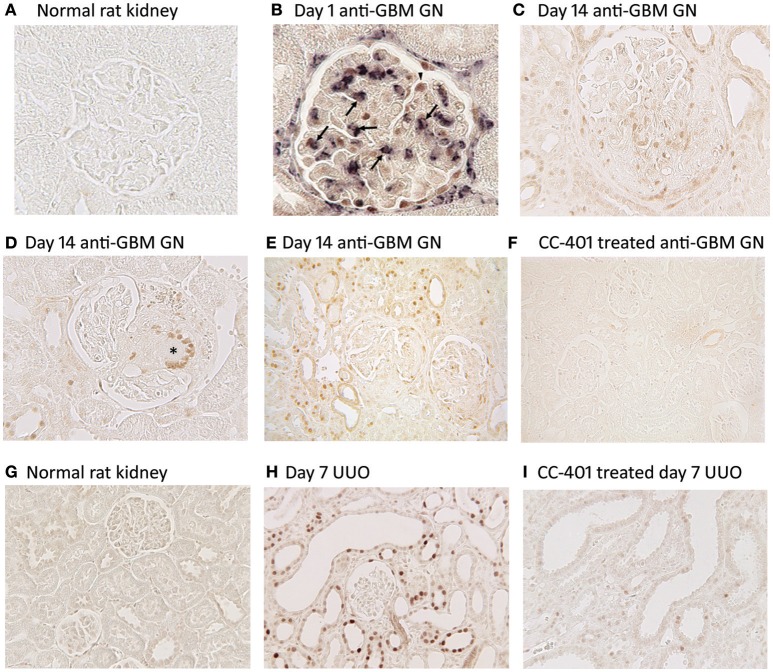
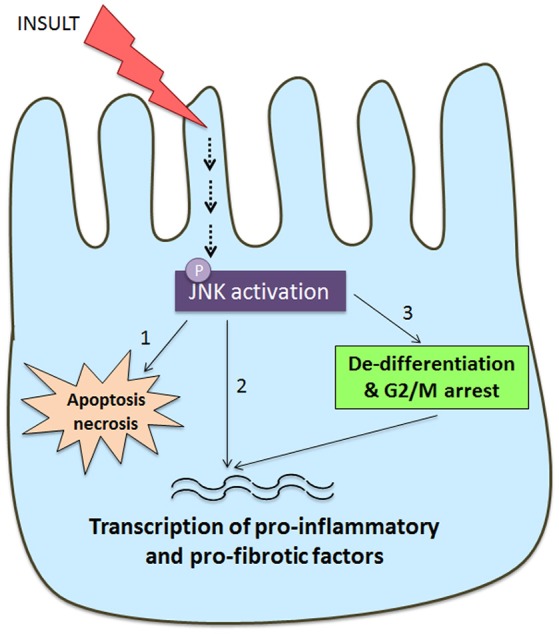
Fibrosis of the glomerular and tubulointerstitial compartments is a common feature of chronic kidney disease leading to end-stage renal failure. This fibrotic process involves a number of pathologic mechanisms, including cell death and inflammation. This review focuses on the role of the c-Jun amino terminal kinase (JNK) signaling pathway in the development of renal fibrosis. The JNK pathway is activated in response to various cellular stresses and plays an important role in cell death and inflammation. Activation of JNK signaling is a common feature in most forms of human kidney injury, evident in both intrinsic glomerular and tubular cells as well as in infiltrating leukocytes. Similar patterns of JNK activation are evident in animal models of acute and chronic renal injury. Administration of JNK inhibitors can protect against acute kidney injury and suppress the development of glomerulosclerosis and tubulointerstitial fibrosis. In particular, JNK activation in tubular epithelial cells may be a pivotal mechanism in determining the outcome of both acute kidney injury and progression of chronic kidney disease. JNK signaling promotes tubular epithelial cell production of pro-inflammatory and pro-fibrotic molecules as well as tubular cell de-differentiation toward a mesenchymal phenotype. However, the role of JNK within renal fibroblasts is less well-characterized. The JNK pathway interacts with other pro-fibrotic pathways, most notable with the TGF-β/SMAD pathway. JNK activation can augment TGF-β gene transcription, induce expression of enzymes that activate the latent form of TGF-β, and JNK directly phosphorylates SMAD3 to enhance transcription of pro-fibrotic molecules. In conclusion, JNK signaling plays an integral role in several key mechanisms operating in renal fibrosis. Targeting of JNK enzymes has therapeutic potential for the treatment of fibrotic kidney diseases.
Keywords: ASK1; SMAD3; apoptosis; kidney disease; kidney inflammation; p38 MAPK.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous