

Control of Phagocytosis by Microbial Pathogens
- PMID: 29114249
- PMCID: PMC5660709
- DOI: 10.3389/fimmu.2017.01368
Control of Phagocytosis by Microbial Pathogens
Abstract
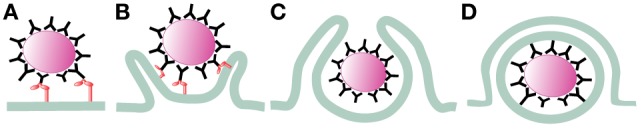
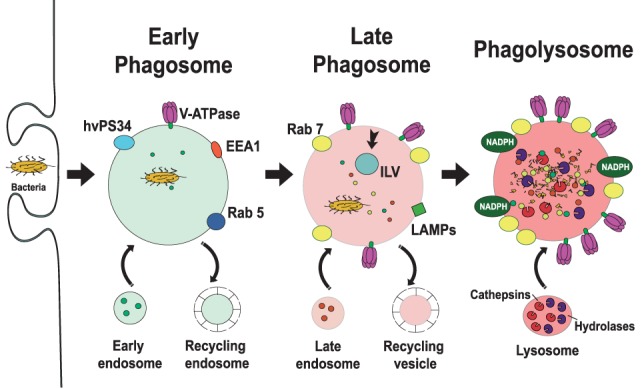
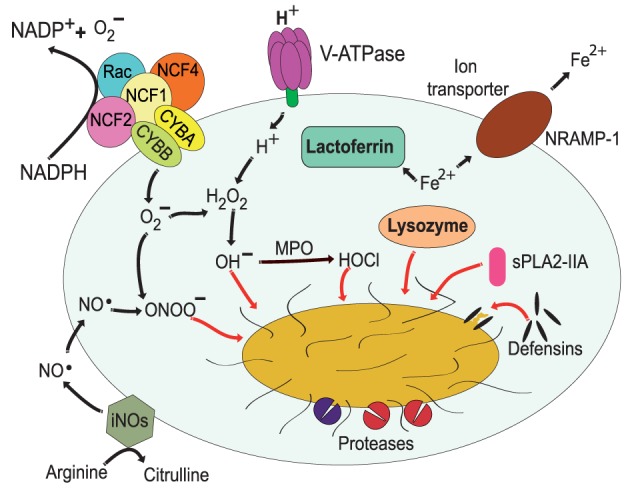
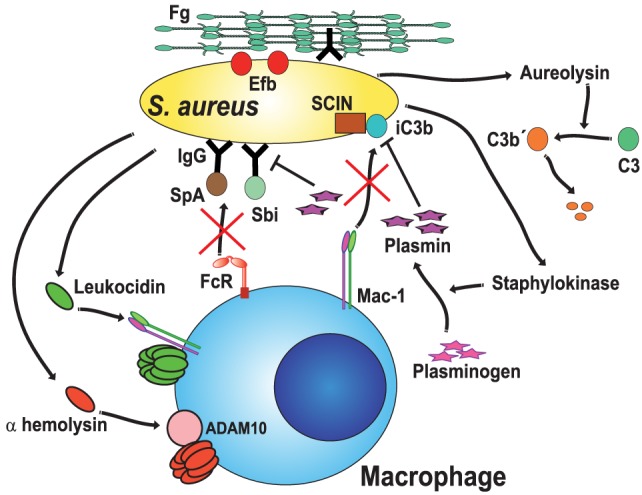
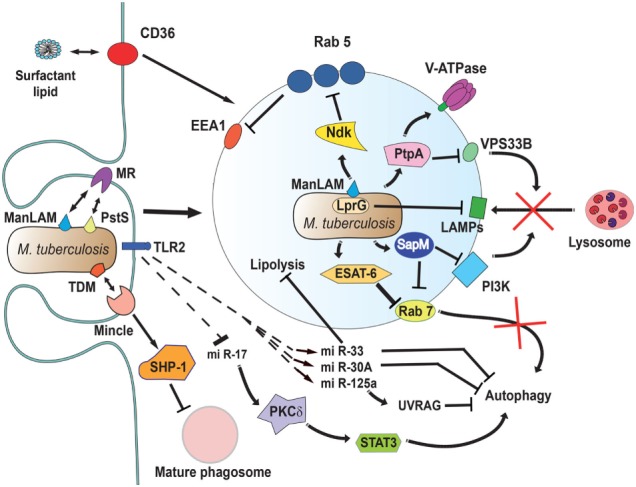
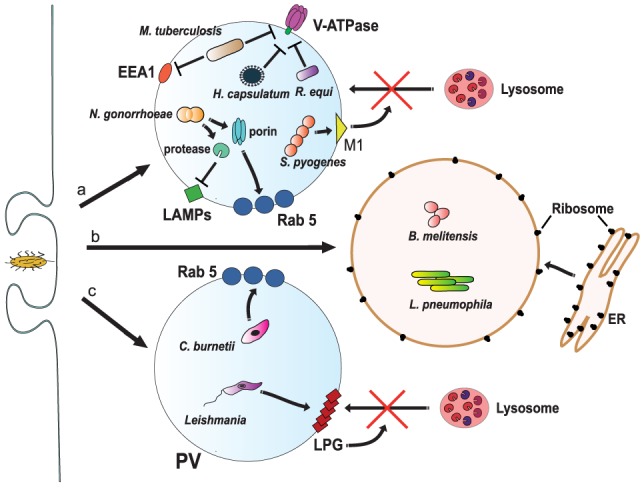
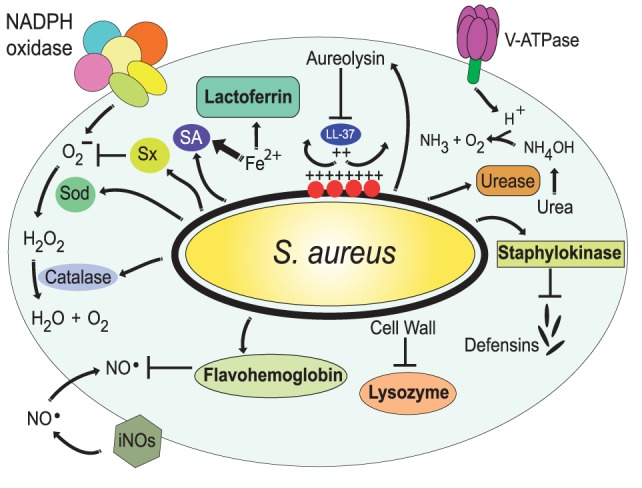
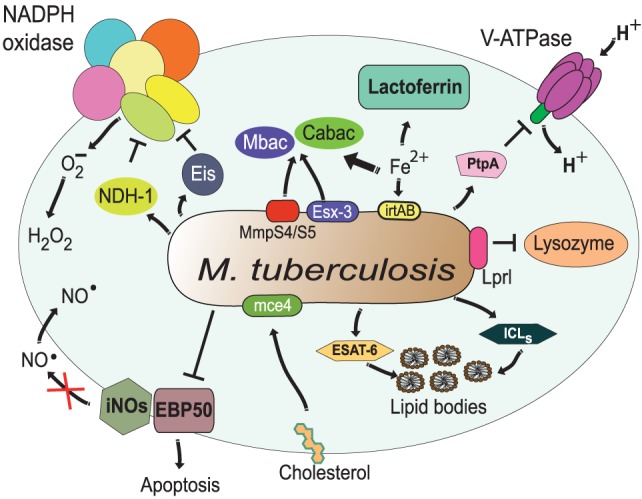
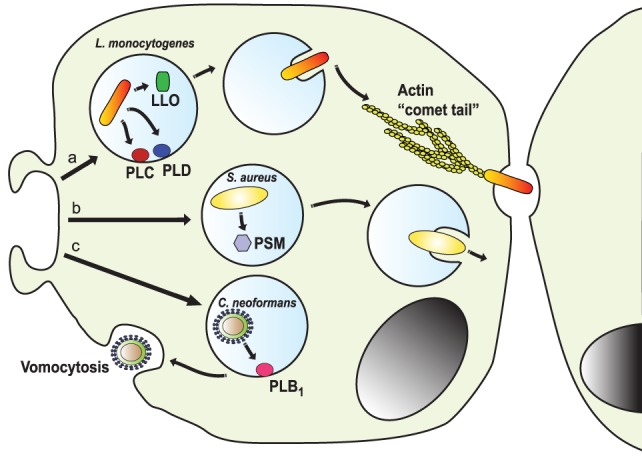
Phagocytosis is a fundamental process of cells to capture and ingest foreign particles. Small unicellular organisms such as free-living amoeba use this process to acquire food. In pluricellular organisms, phagocytosis is a universal phenomenon that all cells are able to perform (including epithelial, endothelial, fibroblasts, etc.), but some specialized cells (such as neutrophils and macrophages) perform this very efficiently and were therefore named professional phagocytes by Rabinovitch. Cells use phagocytosis to capture and clear all particles larger than 0.5 µm, including pathogenic microorganisms and cellular debris. Phagocytosis involves a series of steps from recognition of the target particle, ingestion of it in a phagosome (phagocytic vacuole), maturation of this phagosome into a phagolysosome, to the final destruction of the ingested particle in the robust antimicrobial environment of the phagolysosome. For the most part, phagocytosis is an efficient process that eliminates invading pathogens and helps maintaining homeostasis. However, several pathogens have also evolved different strategies to prevent phagocytosis from proceeding in a normal way. These pathogens have a clear advantage to perpetuate the infection and continue their replication. Here, we present an overview of the phagocytic process with emphasis on the antimicrobial elements professional phagocytes use. We also summarize the current knowledge on the microbial strategies different pathogens use to prevent phagocytosis either at the level of ingestion, phagosome formation, and maturation, and even complete escape from phagosomes.
Keywords: bacteria; infection; inflammation; macrophage; neutrophil; phagolysosome; phagosome maturation.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources