

Comparative genome analysis of programmed DNA elimination in nematodes
- PMID: 29118011
- PMCID: PMC5741062
- DOI: 10.1101/gr.225730.117
Comparative genome analysis of programmed DNA elimination in nematodes
Abstract
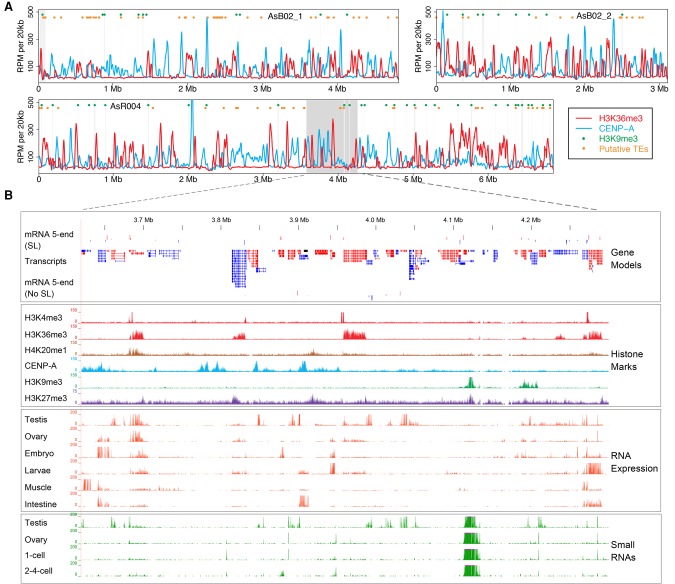
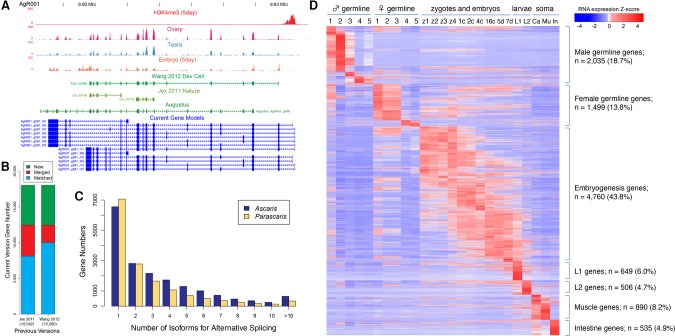
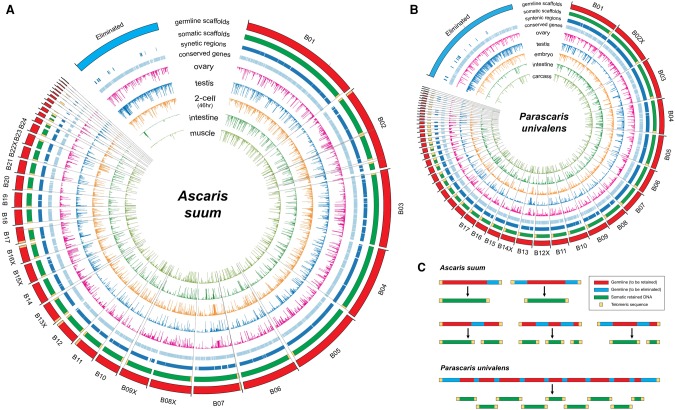
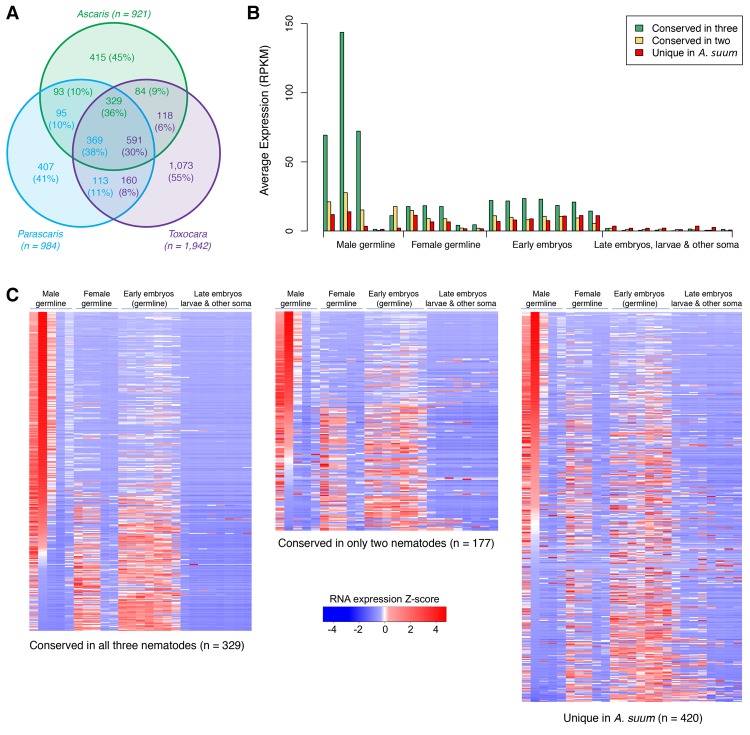
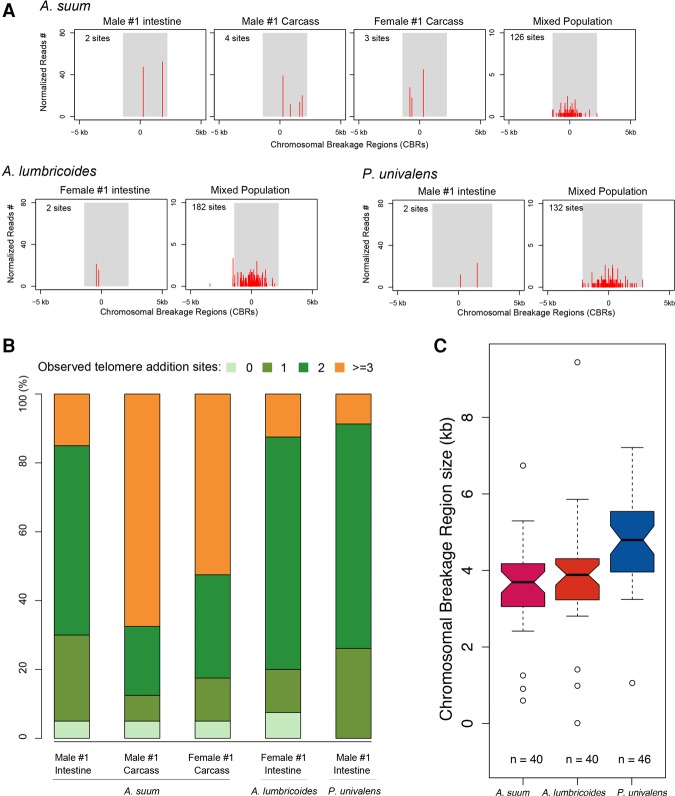
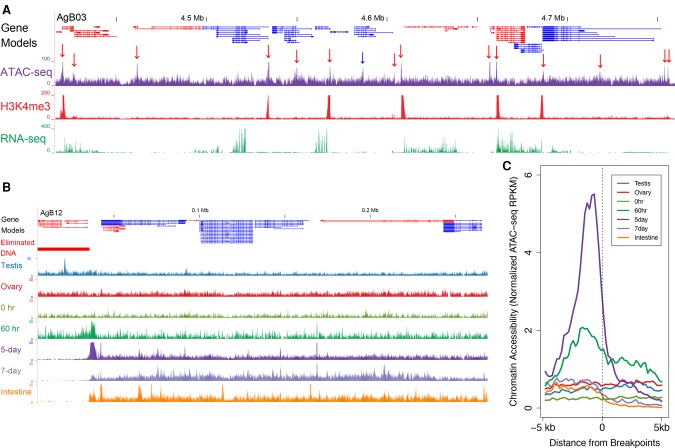
Programmed DNA elimination is a developmentally regulated process leading to the reproducible loss of specific genomic sequences. DNA elimination occurs in unicellular ciliates and a variety of metazoans, including invertebrates and vertebrates. In metazoa, DNA elimination typically occurs in somatic cells during early development, leaving the germline genome intact. Reference genomes for metazoa that undergo DNA elimination are not available. Here, we generated germline and somatic reference genome sequences of the DNA eliminating pig parasitic nematode Ascaris suum and the horse parasite Parascaris univalens. In addition, we carried out in-depth analyses of DNA elimination in the parasitic nematode of humans, Ascaris lumbricoides, and the parasitic nematode of dogs, Toxocara canis. Our analysis of nematode DNA elimination reveals that in all species, repetitive sequences (that differ among the genera) and germline-expressed genes (approximately 1000-2000 or 5%-10% of the genes) are eliminated. Thirty-five percent of these eliminated genes are conserved among these nematodes, defining a core set of eliminated genes that are preferentially expressed during spermatogenesis. Our analysis supports the view that DNA elimination in nematodes silences germline-expressed genes. Over half of the chromosome break sites are conserved between Ascaris and Parascaris, whereas only 10% are conserved in the more divergent T. canis. Analysis of the chromosomal breakage regions suggests a sequence-independent mechanism for DNA breakage followed by telomere healing, with the formation of more accessible chromatin in the break regions prior to DNA elimination. Our genome assemblies and annotations also provide comprehensive resources for analysis of DNA elimination, parasitology research, and comparative nematode genome and epigenome studies.
© 2017 Wang et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
References
-
- Bachmann-Waldmann C, Jentsch S, Tobler H, Muller F. 2004. Chromatin diminution leads to rapid evolutionary changes in the organization of the germ line genomes of the parasitic nematodes A. suum and P. univalens. Mol Biochem Parasitol 134: 53–64. - PubMed
-
- Baird SE, Klobutcher LA. 1989. Characterization of chromosome fragmentation in two protozoans and identification of a candidate fragmentation sequence in Euplotes crassus. Genes Dev 3: 585–597. - PubMed
-
- Bassing CH, Swat W, Alt FW. 2002. The mechanism and regulation of chromosomal V(D)J recombination. Cell 109(Suppl: S45–S55. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases