

Carbon monoxide in lung cell physiology and disease
- PMID: 29118026
- PMCID: PMC5866434
- DOI: 10.1152/ajpcell.00022.2017
Carbon monoxide in lung cell physiology and disease
Abstract
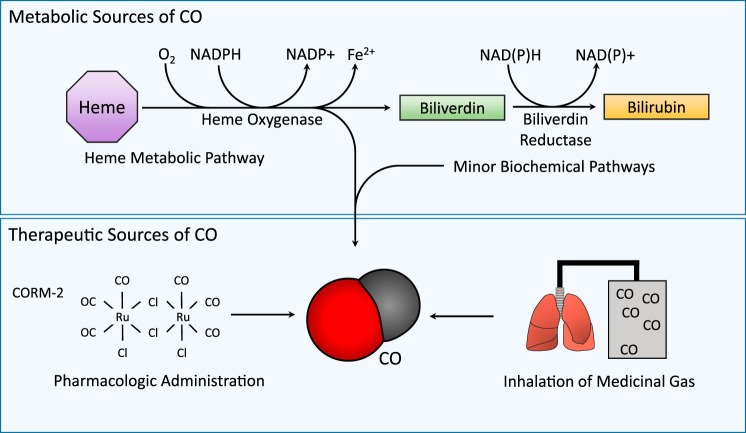
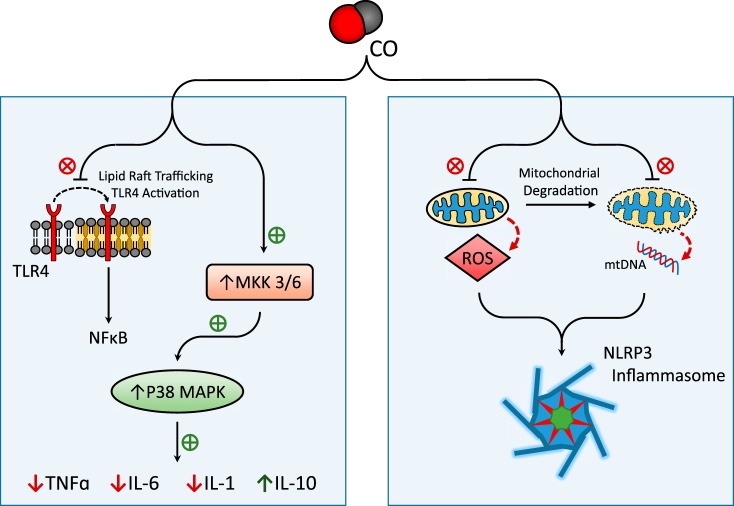
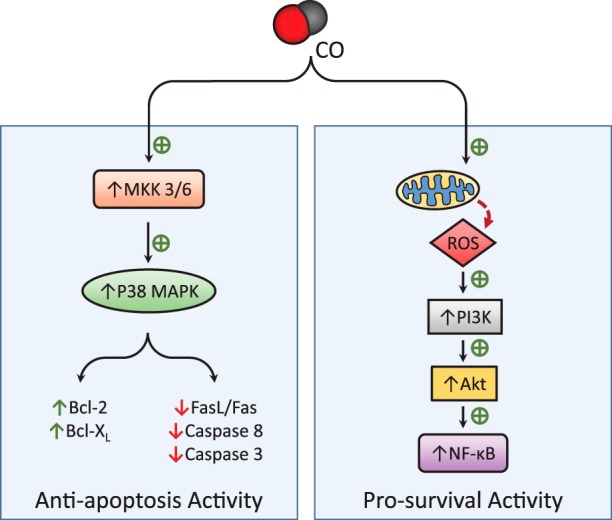
Carbon monoxide (CO) is an endogenously produced gas that has gained recognition as a biological signal transduction effector with properties similar, but not identical, to that of nitric oxide (NO). CO, which binds primarily to heme iron, may activate the hemoprotein guanylate cyclase, although with lower potency than NO. Furthermore, CO can modulate the activities of several cellular signaling molecules such as p38 MAPK, ERK1/2, JNK, Akt, NF-κB, and others. Emerging studies suggest that mitochondria, the energy-generating organelle of cells, represent a key target of CO action in eukaryotes. Dose-dependent modulation of mitochondrial function by CO can result in alteration of mitochondrial membrane potential, mitochondrial reactive oxygen species production, release of proapoptotic and proinflammatory mediators, as well as the inhibition of respiration at high concentration. CO, through modulation of signaling pathways, can impact key biological processes including autophagy, mitochondrial biogenesis, programmed cell death (apoptosis), cellular proliferation, inflammation, and innate immune responses. Inhaled CO is widely known as an inhalation hazard due to its rapid complexation with hemoglobin, resulting in impaired oxygen delivery to tissues and hypoxemia. Despite systemic and cellular toxicity at high concentrations, CO has demonstrated cyto- and tissue-protective effects at low concentration in animal models of organ injury and disease. These include models of acute lung injury (e.g., hyperoxia, hypoxia, ischemia-reperfusion, mechanical ventilation, bleomycin) and sepsis. The success of CO as a candidate therapeutic in preclinical models suggests potential clinical application in inflammatory and proliferative disorders, which is currently under evaluation in clinical trials.
Keywords: carbon monoxide; cell death; cell signaling; inflammation; lung disease; mitochondria; reactive oxygen species; sepsis.
Figures
References
-
- Ahmad S, Hewett PW, Fujisawa T, Sissaoui S, Cai M, Gueron G, Al-Ani B, Cudmore M, Ahmed SF, Wong MK, Wegiel B, Otterbein LE, Vítek L, Ramma W, Wang K, Ahmed A. Carbon monoxide inhibits sprouting angiogenesis and vascular endothelial growth factor receptor-2 phosphorylation. Thromb Haemost 113: 329–337, 2015. doi:10.1160/TH14-01-0002. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous