

The PROSECCO server for chemical shift predictions in ordered and disordered proteins
- PMID: 29119515
- PMCID: PMC5711976
- DOI: 10.1007/s10858-017-0145-2
The PROSECCO server for chemical shift predictions in ordered and disordered proteins
Abstract
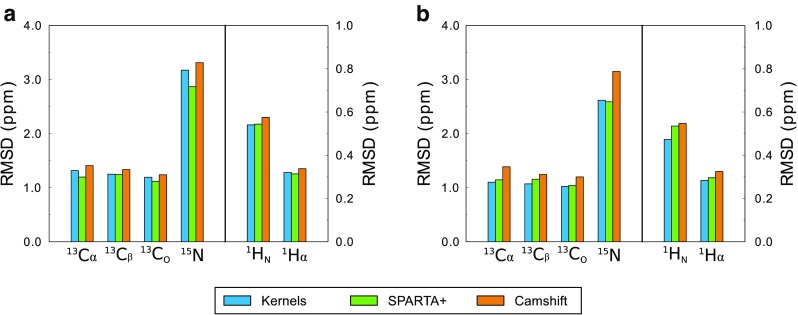
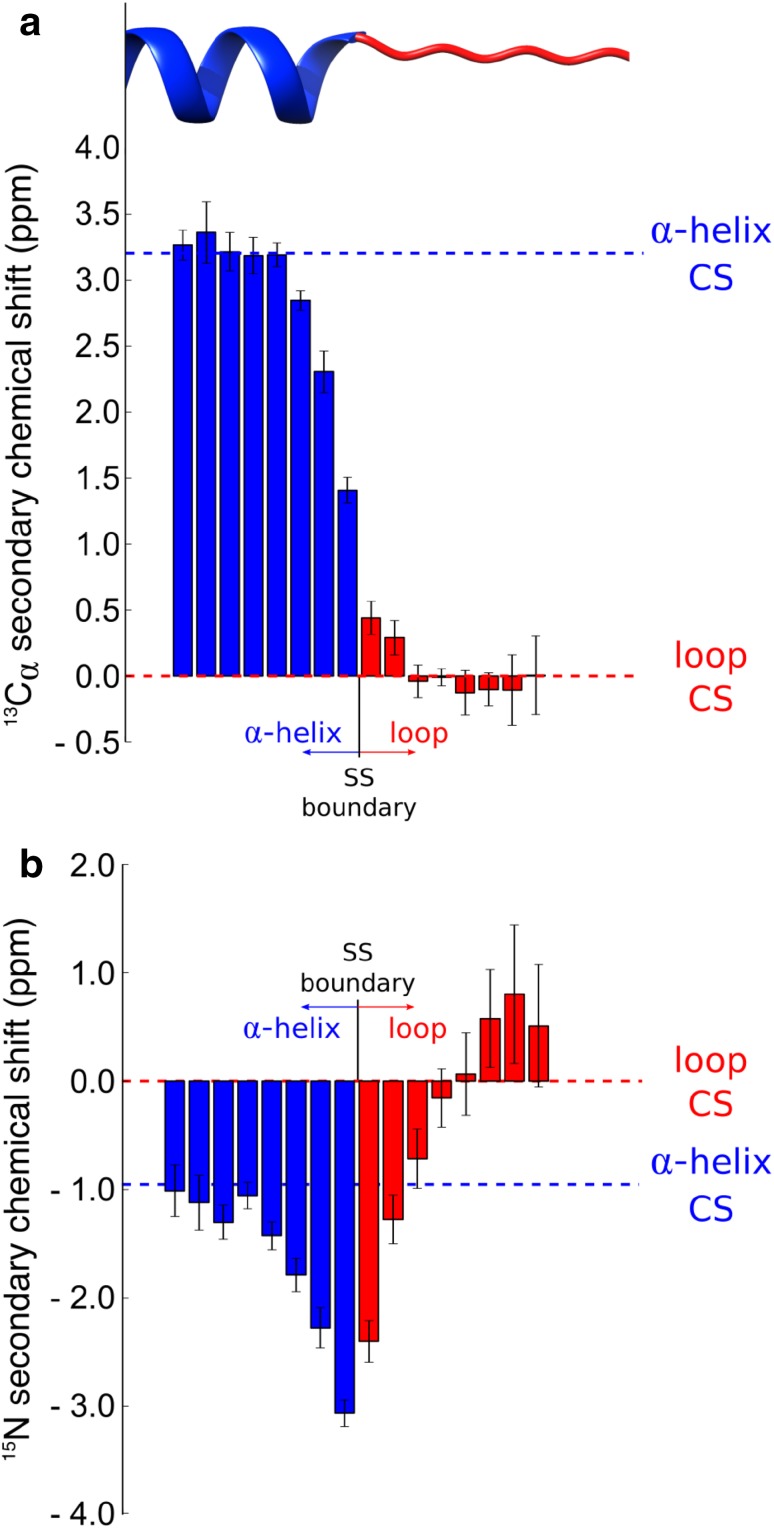
The chemical shifts measured in solution-state and solid-state nuclear magnetic resonance (NMR) are powerful probes of the structure and dynamics of protein molecules. The exploitation of chemical shifts requires methods to correlate these data with the protein structures and sequences. We present here an approach to calculate accurate chemical shifts in both ordered and disordered proteins using exclusively the information contained in their sequences. Our sequence-based approach, protein sequences and chemical shift correlations (PROSECCO), achieves the accuracy of the most advanced structure-based methods in the characterization of chemical shifts of folded proteins and improves the state of the art in the study of disordered proteins. Our analyses revealed fundamental insights on the structural information carried by NMR chemical shifts of structured and unstructured protein states.
Keywords: Biomolecular NMR; Chemical shift predictions; Disordered proteins.
Figures



References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
