

Missing data and technical variability in single-cell RNA-sequencing experiments
- PMID: 29121214
- PMCID: PMC6215955
- DOI: 10.1093/biostatistics/kxx053
Missing data and technical variability in single-cell RNA-sequencing experiments
Abstract
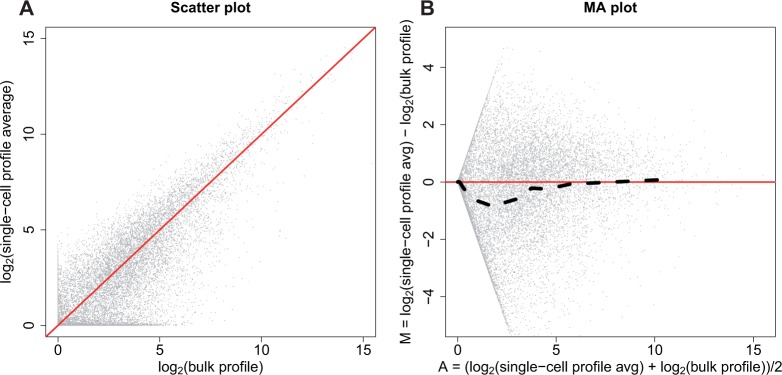
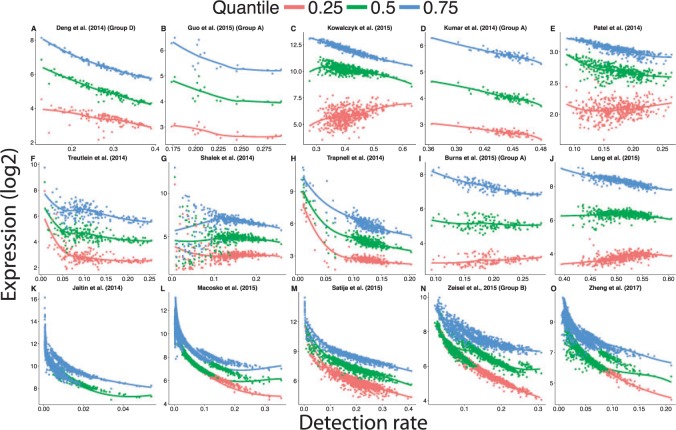
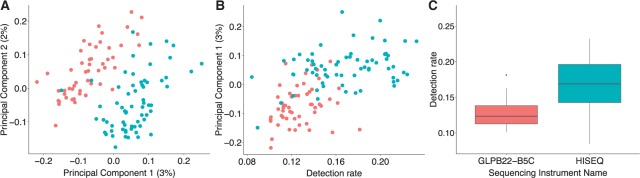
Until recently, high-throughput gene expression technology, such as RNA-Sequencing (RNA-seq) required hundreds of thousands of cells to produce reliable measurements. Recent technical advances permit genome-wide gene expression measurement at the single-cell level. Single-cell RNA-Seq (scRNA-seq) is the most widely used and numerous publications are based on data produced with this technology. However, RNA-seq and scRNA-seq data are markedly different. In particular, unlike RNA-seq, the majority of reported expression levels in scRNA-seq are zeros, which could be either biologically-driven, genes not expressing RNA at the time of measurement, or technically-driven, genes expressing RNA, but not at a sufficient level to be detected by sequencing technology. Another difference is that the proportion of genes reporting the expression level to be zero varies substantially across single cells compared to RNA-seq samples. However, it remains unclear to what extent this cell-to-cell variation is being driven by technical rather than biological variation. Furthermore, while systematic errors, including batch effects, have been widely reported as a major challenge in high-throughput technologies, these issues have received minimal attention in published studies based on scRNA-seq technology. Here, we use an assessment experiment to examine data from published studies and demonstrate that systematic errors can explain a substantial percentage of observed cell-to-cell expression variability. Specifically, we present evidence that some of these reported zeros are driven by technical variation by demonstrating that scRNA-seq produces more zeros than expected and that this bias is greater for lower expressed genes. In addition, this missing data problem is exacerbated by the fact that this technical variation varies cell-to-cell. Then, we show how this technical cell-to-cell variability can be confused with novel biological results. Finally, we demonstrate and discuss how batch-effects and confounded experiments can intensify the problem.
Figures


References
-
- Achim, K., Pettit, J.-B., Saraiva, L. R., Gavriouchkina, D., Larsson, T., Arendt, D. and Marioni, J. C. (2015). High-throughput spatial mapping of single-cell RNA-seq data to tissue of origin. Nature Biotechnology 33, 503–509. - PubMed
-
- Borel, C., Ferreira, P. G., Santoni, F., Delaneau, O., Fort, A., Popadin, K. Y., Garieri, M., Falconnet, E., Ribaux, P., Guipponi, M., Padioleau, I., Carninci, P., Dermitzakis, E. T.. and others (2015). Biased allelic expression in human primary fibroblast single cells. American Journal of Human Genetics 96, 70–80. - PMC - PubMed
-
- Bray, N. L., Pimentel, H., Melsted, P. and Pachter, L. (2016). Near-optimal probabilistic RNA-seq quantification. Nature Biotechnology 34, 525–527. - PubMed
-
- Brennecke, P., Anders, S., Kim, J. K., Kołodziejczyk, A. A., Zhang, X., Proserpio, V., Baying, B., Benes, V., Teichmann, S. A., Marioni, J. C.. and others (2013). Accounting for technical noise in single-cell RNA-seq experiments. Nature Methods 10, 1093–1095. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
