

Meta-analysis of sequence-based association studies across three cattle breeds reveals 25 QTL for fat and protein percentages in milk at nucleotide resolution
- PMID: 29121857
- PMCID: PMC5680815
- DOI: 10.1186/s12864-017-4263-8
Meta-analysis of sequence-based association studies across three cattle breeds reveals 25 QTL for fat and protein percentages in milk at nucleotide resolution
Abstract
Background: Genotyping and whole-genome sequencing data have been generated for hundreds of thousands of cattle. International consortia used these data to compile imputation reference panels that facilitate the imputation of sequence variant genotypes for animals that have been genotyped using dense microarrays. Association studies with imputed sequence variant genotypes allow for the characterization of quantitative trait loci (QTL) at nucleotide resolution particularly when individuals from several breeds are included in the mapping populations.
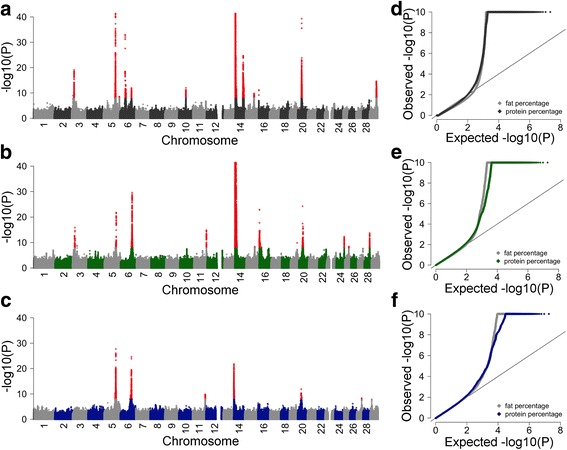
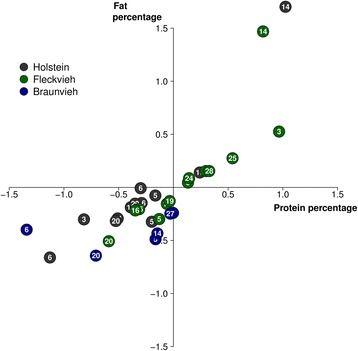
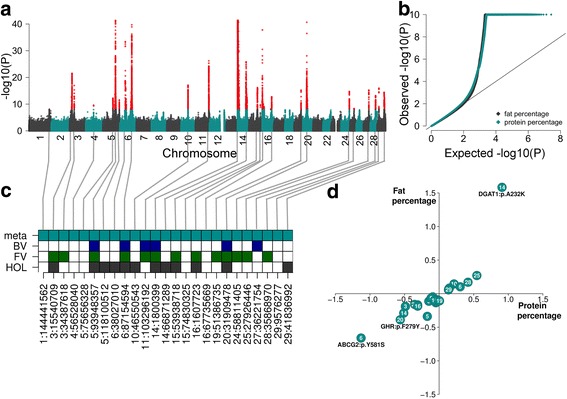
Results: We imputed genotypes for 28 million sequence variants in 17,229 cattle of the Braunvieh, Fleckvieh and Holstein breeds in order to compile large mapping populations that provide high power to identify QTL for milk production traits. Association tests between imputed sequence variant genotypes and fat and protein percentages in milk uncovered between six and thirteen QTL (P < 1e-8) per breed. Eight of the detected QTL were significant in more than one breed. We combined the results across breeds using meta-analysis and identified a total of 25 QTL including six that were not significant in the within-breed association studies. Two missense mutations in the ABCG2 (p.Y581S, rs43702337, P = 4.3e-34) and GHR (p.F279Y, rs385640152, P = 1.6e-74) genes were the top variants at QTL on chromosomes 6 and 20. Another known causal missense mutation in the DGAT1 gene (p.A232K, rs109326954, P = 8.4e-1436) was the second top variant at a QTL on chromosome 14 but its allelic substitution effects were inconsistent across breeds. It turned out that the conflicting allelic substitution effects resulted from flaws in the imputed genotypes due to the use of a multi-breed reference population for genotype imputation.
Conclusions: Many QTL for milk production traits segregate across breeds and across-breed meta-analysis has greater power to detect such QTL than within-breed association testing. Association testing between imputed sequence variant genotypes and phenotypes of interest facilitates identifying causal mutations provided the accuracy of imputation is high. However, true causal mutations may remain undetected when the imputed sequence variant genotypes contain flaws. It is highly recommended to validate the effect of known causal variants in order to assess the ability to detect true causal mutations in association studies with imputed sequence variants.
Keywords: Cattle; Dairy traits; Meta-analysis; Qtl; Sequence imputation.
Conflict of interest statement
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
References
-
- Goddard ME, Hayes BJ. Genomic selection based on dense genotypes inferred from sparse genotypes. Proc Adv Anim Breed Genet. 2009;18:26–29.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
