

A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys
- PMID: 29121992
- PMCID: PMC5679155
- DOI: 10.1186/s13395-017-0141-y
A mouse anti-myostatin antibody increases muscle mass and improves muscle strength and contractility in the mdx mouse model of Duchenne muscular dystrophy and its humanized equivalent, domagrozumab (PF-06252616), increases muscle volume in cynomolgus monkeys
Abstract
Background: The treatments currently approved for Duchenne muscular dystrophy (DMD), a progressive skeletal muscle wasting disease, address the needs of only a small proportion of patients resulting in an urgent need for therapies that benefit all patients regardless of the underlying mutation. Myostatin is a member of the transforming growth factor-β (TGF-β) family of ligands and is a negative regulator of skeletal muscle mass. Loss of myostatin has been shown to increase muscle mass and improve muscle function in both normal and dystrophic mice. Therefore, myostatin blockade via a specific antibody could ameliorate the muscle weakness in DMD patients by increasing skeletal muscle mass and function, thereby reducing patients' functional decline.
Methods: A murine anti-myostatin antibody, mRK35, and its humanized analog, domagrozumab, were developed and their ability to inhibit several TGB-β ligands was measured using a cell-based Smad-activity reporter system. Normal and mdx mice were treated with mRK35 to examine the antibody's effect on body weight, lean mass, muscle weights, grip strength, ex vivo force production, and fiber size. The humanized analog (domagrozumab) was tested in non-human primates (NHPs) for changes in skeletal muscle mass and volume as well as target engagement via modulation of circulating myostatin.
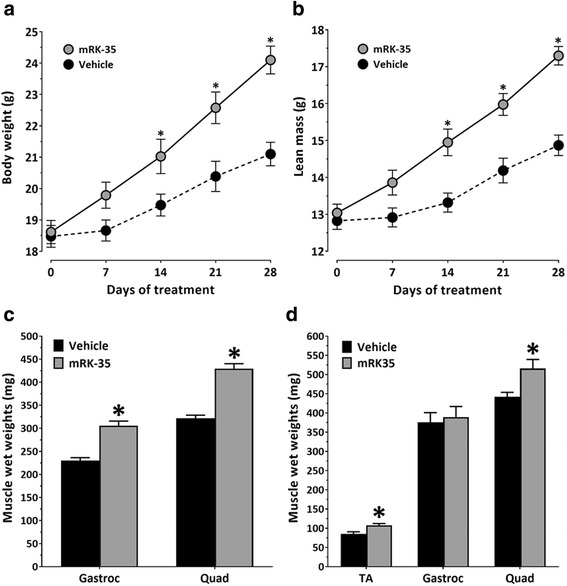
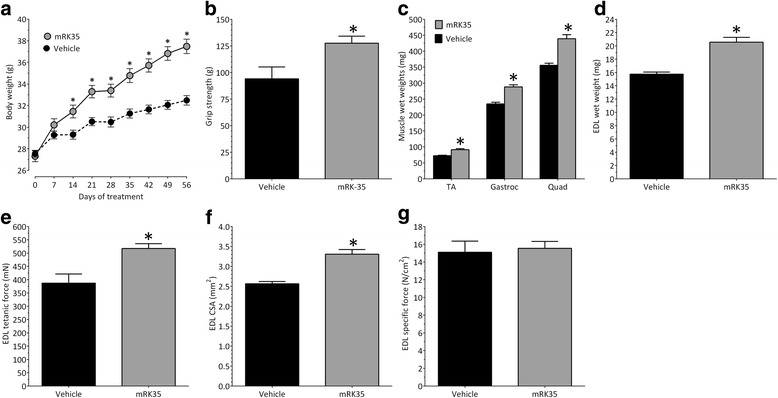
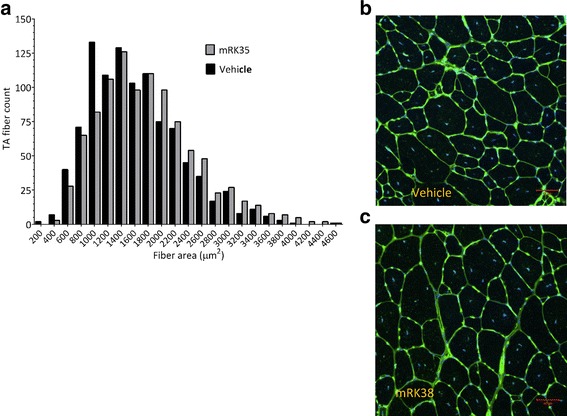
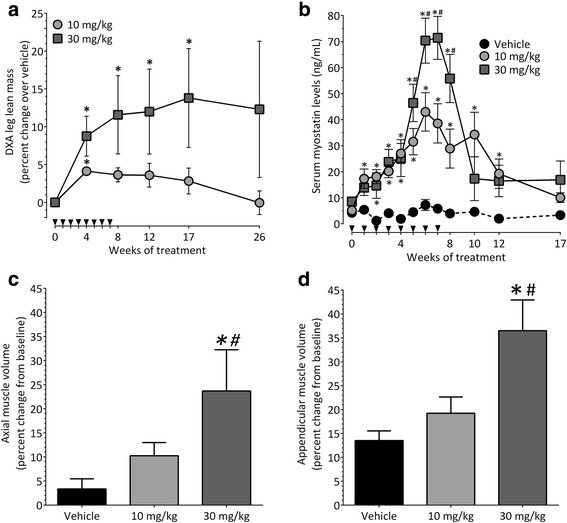
Results: Both the murine and human antibodies are specific and potent inhibitors of myostatin and GDF11. mRK35 is able to increase body weight, lean mass, and muscle weights in normal mice. In mdx mice, mRK35 significantly increased body weight, muscle weights, grip strength, and ex vivo force production in the extensor digitorum longus (EDL) muscle. Further, tibialis anterior (TA) fiber size was significantly increased. NHPs treated with domagrozumab demonstrated a dose-dependent increase in lean mass and muscle volume and exhibited increased circulating levels of myostatin demonstrating target engagement.
Conclusions: We demonstrated that the potent anti-myostatin antibody mRK35 and its clinical analog, domagrozumab, were able to induce muscle anabolic activity in both rodents, including the mdx mouse model of DMD, and non-human primates. A Phase 2, potentially registrational, clinical study with domagrozumab in DMD patients is currently underway.
Keywords: Duchenne muscular dystrophy; Hypertrophy; Monoclonal antibody; Myostatin; Skeletal muscle; mdx.
Conflict of interest statement
Ethics approval
All animal procedures were approved by the IACUC and were carried out in an AAALAC accredited facility
Consent for publication
Not applicable
Competing interests
All authors were employees of Pfizer when these studies were conducted
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Figures
Similar articles
-
A GDF11/myostatin inhibitor, GDF11 propeptide-Fc, increases skeletal muscle mass and improves muscle strength in dystrophic mdx mice.Skelet Muscle. 2019 May 27;9(1):16. doi: 10.1186/s13395-019-0197-y. Skelet Muscle. 2019. PMID: 31133057 Free PMC article.
-
Pre-clinical evaluation of N-acetylcysteine reveals side effects in the mdx mouse model of Duchenne muscular dystrophy.J Physiol. 2017 Dec 1;595(23):7093-7107. doi: 10.1113/JP274229. Epub 2017 Sep 30. J Physiol. 2017. PMID: 28887840 Free PMC article.
-
Contractile efficiency of dystrophic mdx mouse muscle: in vivo and ex vivo assessment of adaptation to exercise of functional end points.J Appl Physiol (1985). 2017 Apr 1;122(4):828-843. doi: 10.1152/japplphysiol.00776.2015. Epub 2017 Jan 5. J Appl Physiol (1985). 2017. PMID: 28057817
-
Interference with myostatin/ActRIIB signaling as a therapeutic strategy for Duchenne muscular dystrophy.Curr Gene Ther. 2012 Jun;12(3):245-59. doi: 10.2174/156652312800840577. Curr Gene Ther. 2012. PMID: 22554312 Review.
-
Humanization of the mdx Mouse Phenotype for Duchenne Muscular Dystrophy Modeling: A Metabolic Perspective.J Neuromuscul Dis. 2023;10(6):1003-1012. doi: 10.3233/JND-230126. J Neuromuscul Dis. 2023. PMID: 37574742 Free PMC article. Review.
Cited by
-
Pharmacological Inhibition of Myostatin in a Mouse Model of Typical Nemaline Myopathy Increases Muscle Size and Force.Int J Mol Sci. 2023 Oct 12;24(20):15124. doi: 10.3390/ijms242015124. Int J Mol Sci. 2023. PMID: 37894805 Free PMC article.
-
Structural basis of specific inhibition of extracellular activation of pro- or latent myostatin by the monoclonal antibody SRK-015.J Biol Chem. 2020 Apr 17;295(16):5404-5418. doi: 10.1074/jbc.RA119.012293. Epub 2020 Feb 19. J Biol Chem. 2020. PMID: 32075906 Free PMC article.
-
The Failed Clinical Story of Myostatin Inhibitors against Duchenne Muscular Dystrophy: Exploring the Biology behind the Battle.Cells. 2020 Dec 10;9(12):2657. doi: 10.3390/cells9122657. Cells. 2020. PMID: 33322031 Free PMC article.
-
Quantitative magnetic resonance imaging measures as biomarkers of disease progression in boys with Duchenne muscular dystrophy: a phase 2 trial of domagrozumab.J Neurol. 2022 Aug;269(8):4421-4435. doi: 10.1007/s00415-022-11084-0. Epub 2022 Apr 8. J Neurol. 2022. PMID: 35396602 Free PMC article. Clinical Trial.
-
Bioanalytical methods in doping controls: a review.Bioanalysis. 2025 Mar;17(5):359-370. doi: 10.1080/17576180.2025.2460951. Epub 2025 Feb 7. Bioanalysis. 2025. PMID: 39916648 Review.
References
-
- US Food and Drug Administration. EXONDYS 51 full prescribing information 2016. doi. http://www.accessdata.fda.gov/drugsatfda_docs/label/2016/206488lbl.pdf. Accessed 17 Mar 2017.
-
- McDonald CM, Campbell C, Torricelli RE, Finkel RS, Flanigan KM, Goemans N, Heydemann P, Kaminska A, Kirschner J, Muntoni F0, Osorio AN, Schara U, Sejersen T, Shieh PB, Sweeney HL, Topaloglu H, Tulinius M, Vilchez JJ, Voit T, Wong B, Elfring G, Kroger H, Luo X, McIntosh J, Ong T, Riebling P, Souza M, Spiegel RJ, Peltz SW, Mercuri E; Alfano LN, Eagle M, James MK, Lowes L, Mayhew A, Mazzone ES, Nelson L, Rose KJ, Abdel-Hamid HZ, Apkon SD, Barohn RJ, Bertini E, Bloetzer C, de Vaud LC, Butterfield RJ, Chabrol B, Chae JH, Jongno-Gu DR, Comi GP, Darras BT, Dastgir J, Desguerre I, Escobar RG, Finanger E, Guglieri M, Hughes I, Iannaccone ST, Jones KJ, Karachunski P, Kudr M, Lotze T, Mah JK, Mathews K, Nevo Y, Parsons J, Péréon Y, de Queiroz Campos Araujo AP, Renfroe JB, de Resende MBD, Ryan M, Selby K, Tennekoon G, Vita G. Ataluren in patients with nonsense mutation Duchenne muscular dystrophy (ACT DMD): a multicentre, randomised, double-blind, placebo-controlled, phase 3 trial. Lancet. 2017;390(10101):1489–498. - PubMed
-
- Sarepta Therapeutics. Clinical trials: the basics and our clinical trials: a double-blind, placebo-controlled, multi-center study with an open-label extension to evaluate the efficacy and safety of SRP-4045 and SRP-4053 in patients with Duchenne muscular dystrophy 2017;2017.
-
- ClinicalTrials.gov. Registry of Translarna (Ataluren) in nonsense mutation Duchenne muscular dystrophy: NCT02369731. 2017. doi. https://clinicaltrials.gov/ct2/show/NCT02369731. Accessed 17 Mar 2017.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
