

Prevalence and detection of low-allele-fraction variants in clinical cancer samples
- PMID: 29123093
- PMCID: PMC5680209
- DOI: 10.1038/s41467-017-01470-y
Prevalence and detection of low-allele-fraction variants in clinical cancer samples
Abstract
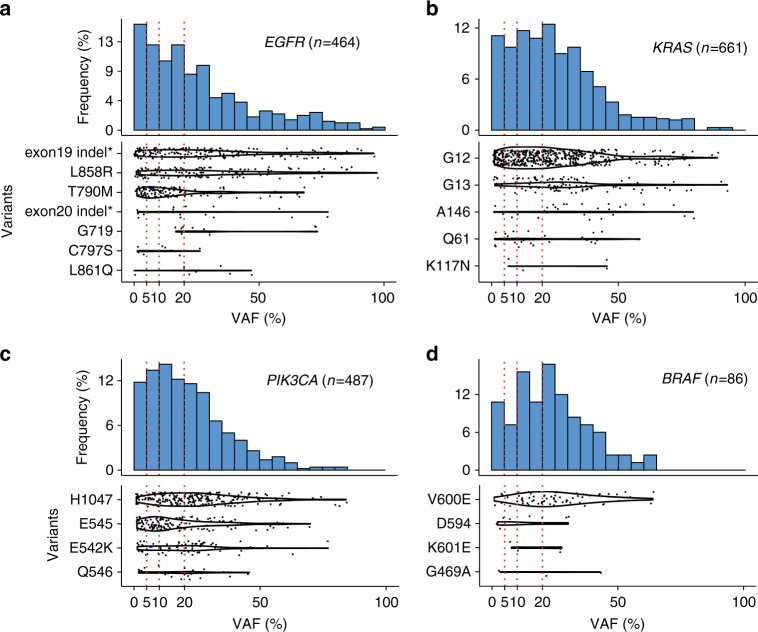
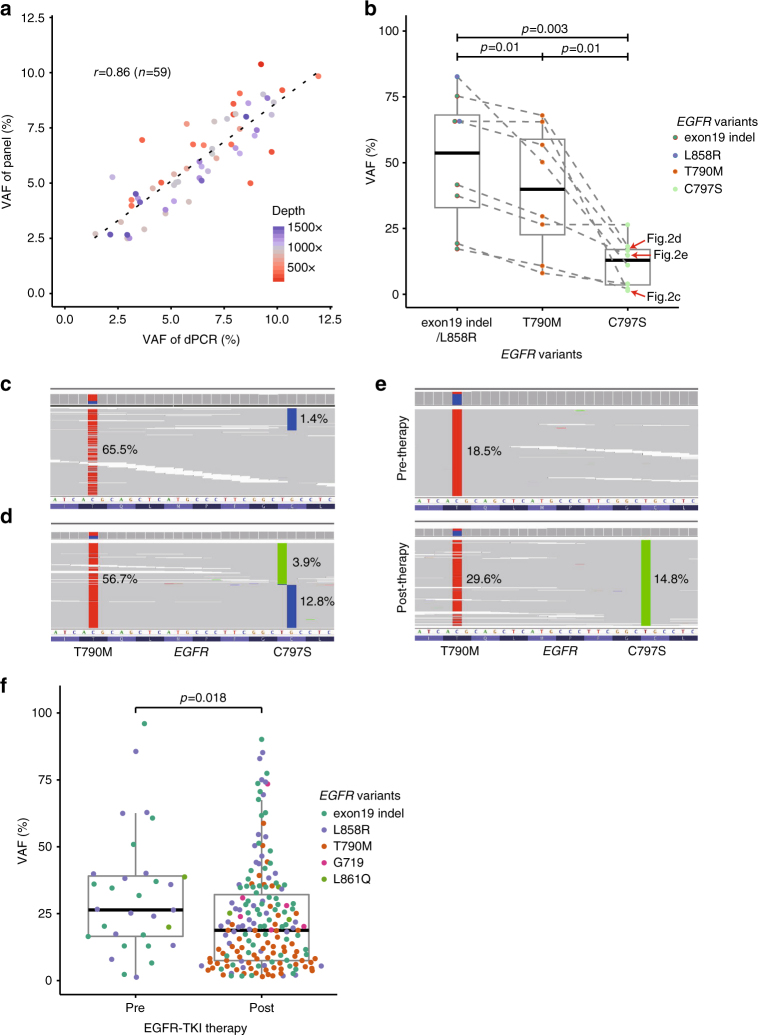
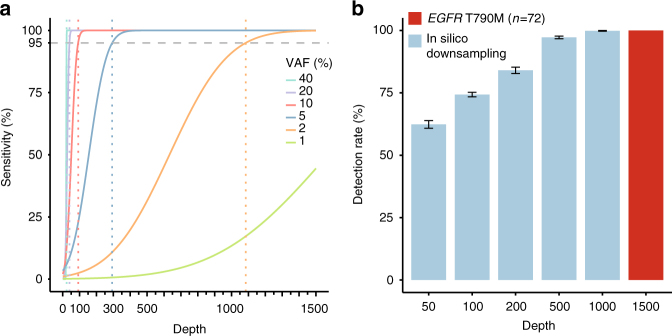
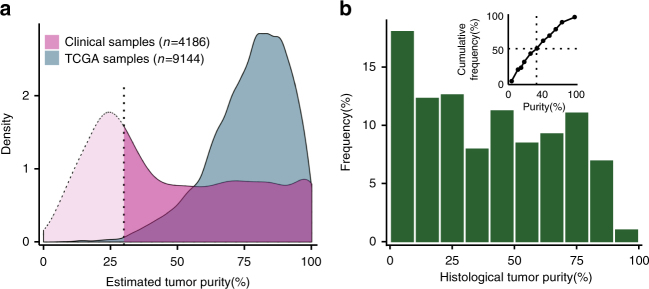
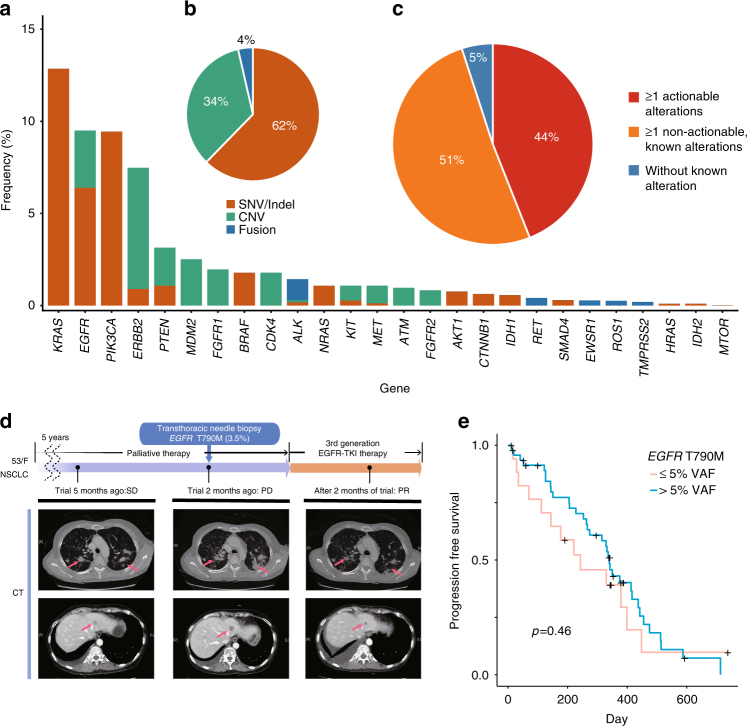
Accurate detection of genomic alterations using high-throughput sequencing is an essential component of precision cancer medicine. We characterize the variant allele fractions (VAFs) of somatic single nucleotide variants and indels across 5095 clinical samples profiled using a custom panel, CancerSCAN. Our results demonstrate that a significant fraction of clinically actionable variants have low VAFs, often due to low tumor purity and treatment-induced mutations. The percentages of mutations under 5% VAF across hotspots in EGFR, KRAS, PIK3CA, and BRAF are 16%, 11%, 12%, and 10%, respectively, with 24% for EGFR T790M and 17% for PIK3CA E545. For clinical relevance, we describe two patients for whom targeted therapy achieved remission despite low VAF mutations. We also characterize the read depths necessary to achieve sensitivity and specificity comparable to current laboratory assays. These results show that capturing low VAF mutations at hotspots by sufficient sequencing coverage and carefully tuned algorithms is imperative for a clinical assay.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
Similar articles
-
Next‑generation sequencing‑based detection of EGFR, KRAS, BRAF, NRAS, PIK3CA, Her‑2 and TP53 mutations in patients with non‑small cell lung cancer.Mol Med Rep. 2018 Aug;18(2):2191-2197. doi: 10.3892/mmr.2018.9210. Epub 2018 Jun 22. Mol Med Rep. 2018. PMID: 29956783 Free PMC article.
-
Consistency and reproducibility of next-generation sequencing and other multigene mutational assays: A worldwide ring trial study on quantitative cytological molecular reference specimens.Cancer Cytopathol. 2017 Aug;125(8):615-626. doi: 10.1002/cncy.21868. Epub 2017 May 5. Cancer Cytopathol. 2017. PMID: 28475299
-
Assessment of the clinical application of detecting EGFR, KRAS, PIK3CA and BRAF mutations in patients with non-small cell lung cancer using next-generation sequencing.Scand J Clin Lab Invest. 2016 Sep;76(5):386-92. doi: 10.1080/00365513.2016.1183813. Epub 2016 May 23. Scand J Clin Lab Invest. 2016. PMID: 27215271
-
A new generation of companion diagnostics: cobas BRAF, KRAS and EGFR mutation detection tests.Expert Rev Mol Diagn. 2014 Jun;14(5):517-24. doi: 10.1586/14737159.2014.910120. Expert Rev Mol Diagn. 2014. PMID: 24844134 Review.
-
Variant allele frequency: a decision-making tool in precision oncology?Trends Cancer. 2023 Dec;9(12):1058-1068. doi: 10.1016/j.trecan.2023.08.011. Epub 2023 Sep 12. Trends Cancer. 2023. PMID: 37704501 Review.
Cited by
-
Mutational Profile and Clonal Evolution of Relapsed/Refractory Diffuse Large B-Cell Lymphoma.Front Oncol. 2021 Mar 11;11:628807. doi: 10.3389/fonc.2021.628807. eCollection 2021. Front Oncol. 2021. PMID: 33777778 Free PMC article.
-
Targeted Sequencing of Plasma-Derived vs. Urinary cfDNA from Patients with Triple-Negative Breast Cancer.Cancers (Basel). 2022 Aug 24;14(17):4101. doi: 10.3390/cancers14174101. Cancers (Basel). 2022. PMID: 36077638 Free PMC article.
-
Development and evaluation of ActSeq: A targeted next-generation sequencing panel for clinical oncology use.PLoS One. 2022 Apr 21;17(4):e0266914. doi: 10.1371/journal.pone.0266914. eCollection 2022. PLoS One. 2022. PMID: 35446881 Free PMC article.
-
Treatment with immune checkpoint inhibitors after EGFR-TKIs in EGFR-mutated lung cancer.Thorac Cancer. 2022 Feb;13(3):386-393. doi: 10.1111/1759-7714.14267. Epub 2021 Dec 13. Thorac Cancer. 2022. PMID: 34904383 Free PMC article.
-
Error-corrected ultradeep next-generation sequencing for detection of clonal haematopoiesis and haematological neoplasms - sensitivity, specificity and accuracy.PLoS One. 2025 Feb 26;20(2):e0318300. doi: 10.1371/journal.pone.0318300. eCollection 2025. PLoS One. 2025. PMID: 40009600 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
