

DNA methylation at enhancers identifies distinct breast cancer lineages
- PMID: 29123100
- PMCID: PMC5680222
- DOI: 10.1038/s41467-017-00510-x
DNA methylation at enhancers identifies distinct breast cancer lineages
Abstract
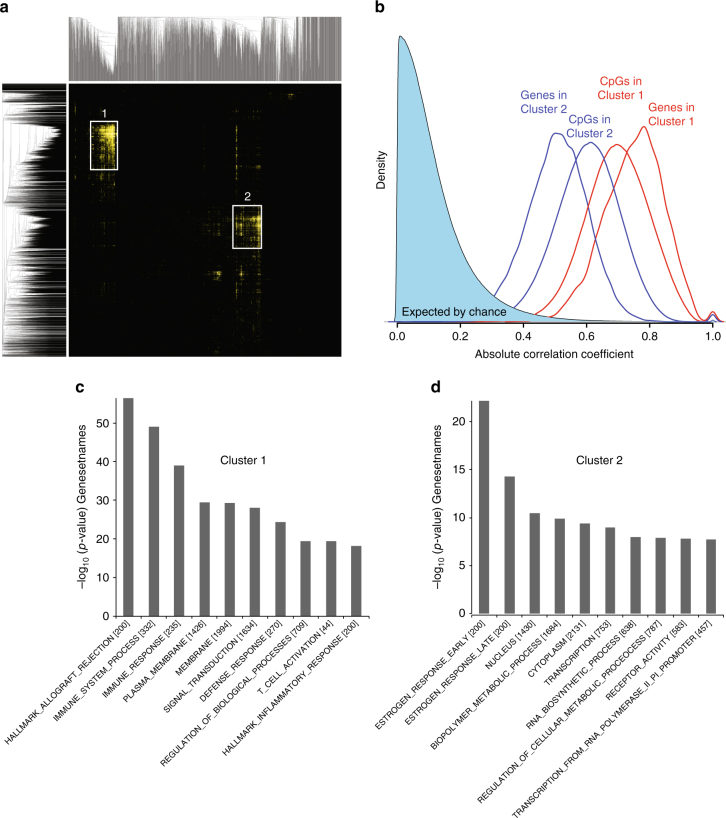
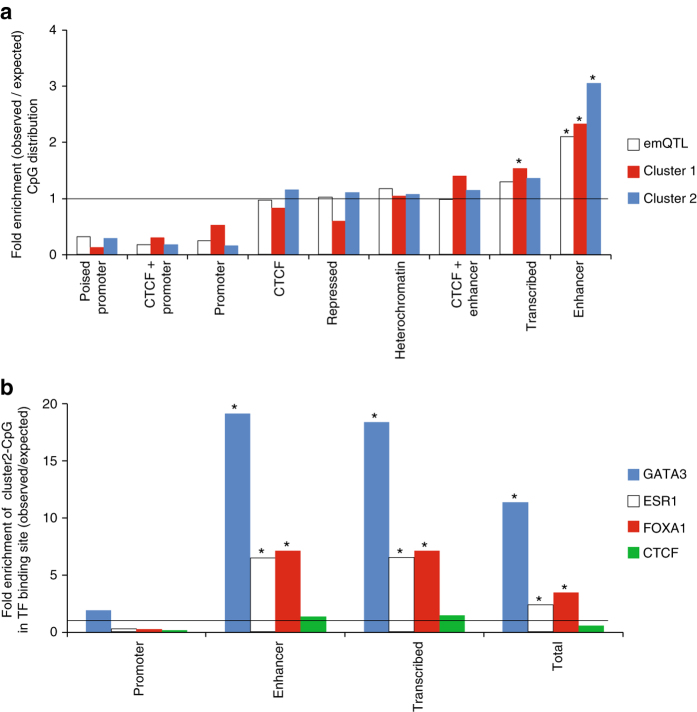
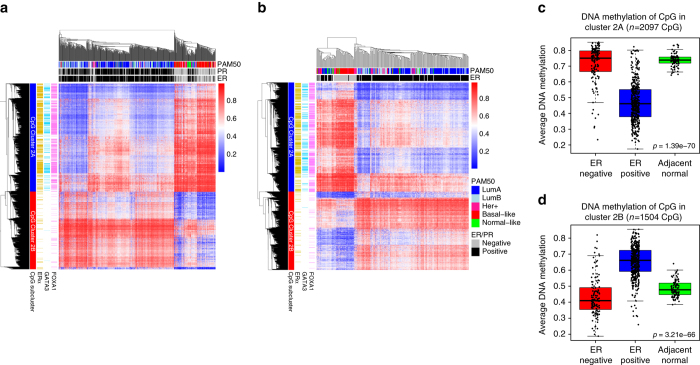
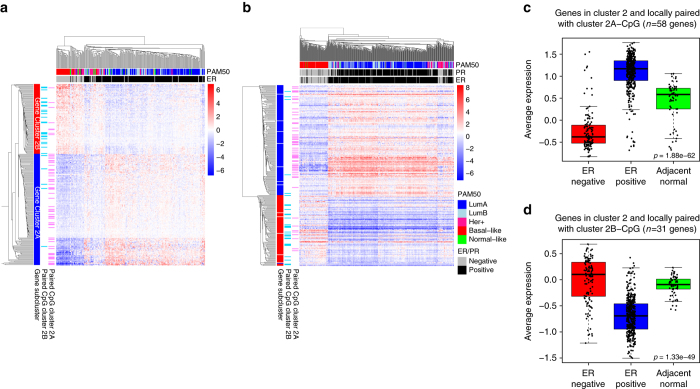
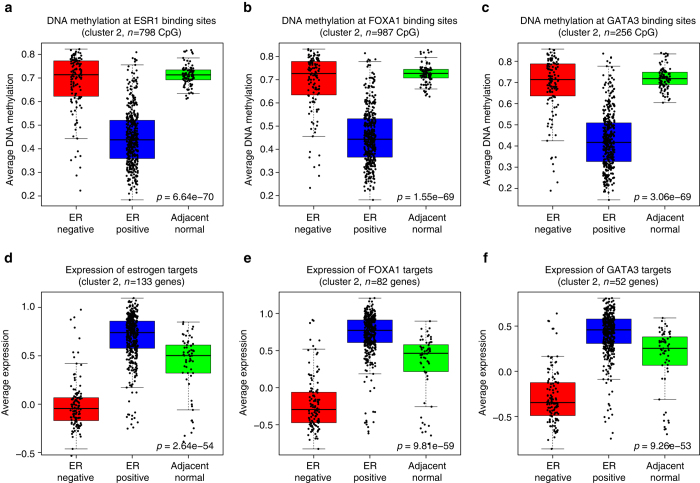
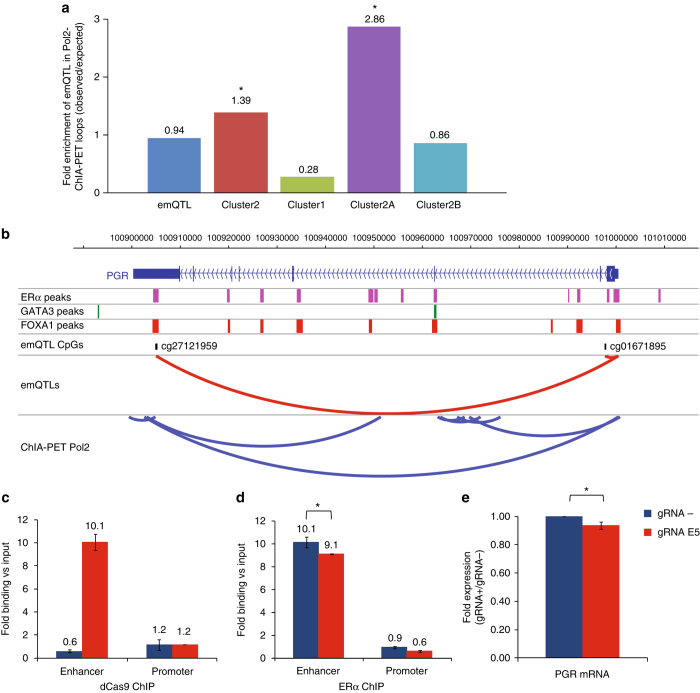
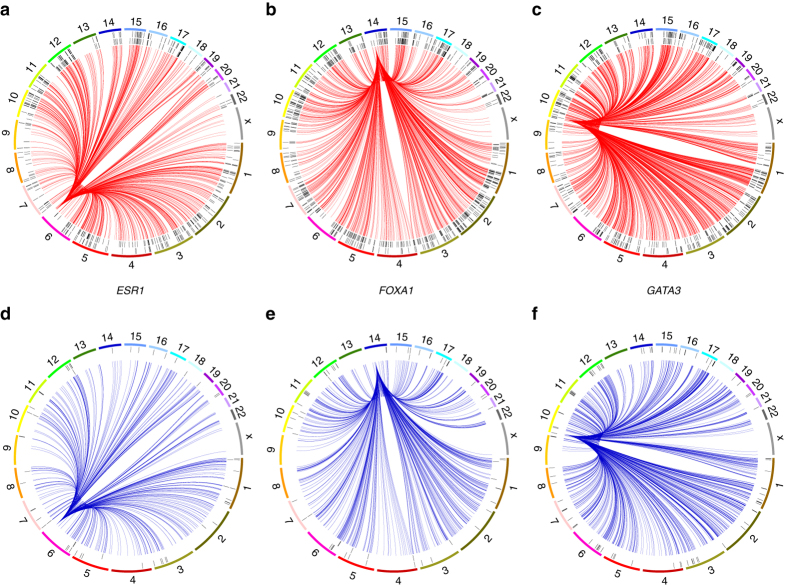
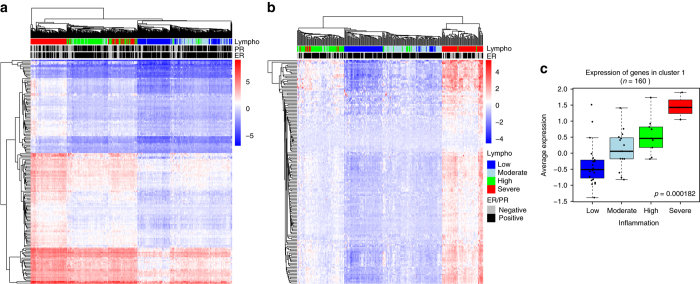
Breast cancers exhibit genome-wide aberrant DNA methylation patterns. To investigate how these affect the transcriptome and which changes are linked to transformation or progression, we apply genome-wide expression-methylation quantitative trait loci (emQTL) analysis between DNA methylation and gene expression. On a whole genome scale, in cis and in trans, DNA methylation and gene expression have remarkably and reproducibly conserved patterns of association in three breast cancer cohorts (n = 104, n = 253 and n = 277). The expression-methylation quantitative trait loci associations form two main clusters; one relates to tumor infiltrating immune cell signatures and the other to estrogen receptor signaling. In the estrogen related cluster, using ChromHMM segmentation and transcription factor chromatin immunoprecipitation sequencing data, we identify transcriptional networks regulated in a cell lineage-specific manner by DNA methylation at enhancers. These networks are strongly dominated by ERα, FOXA1 or GATA3 and their targets were functionally validated using knockdown by small interfering RNA or GRO-seq analysis after transcriptional stimulation with estrogen.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
