

Discovering the stacking landscape of a pyridine-pyridine system
- PMID: 29124340
- PMCID: PMC5680376
- DOI: 10.1007/s00894-017-3496-4
Discovering the stacking landscape of a pyridine-pyridine system
Abstract
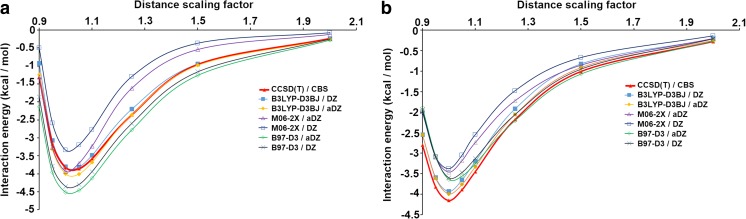
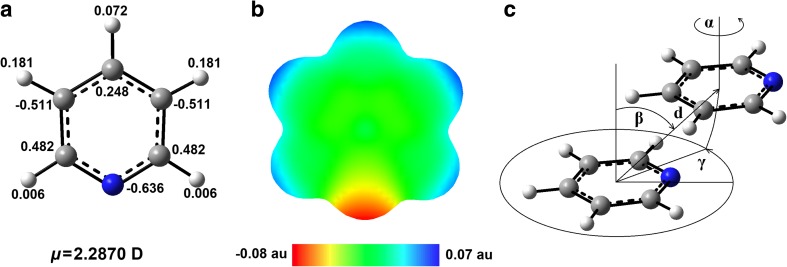
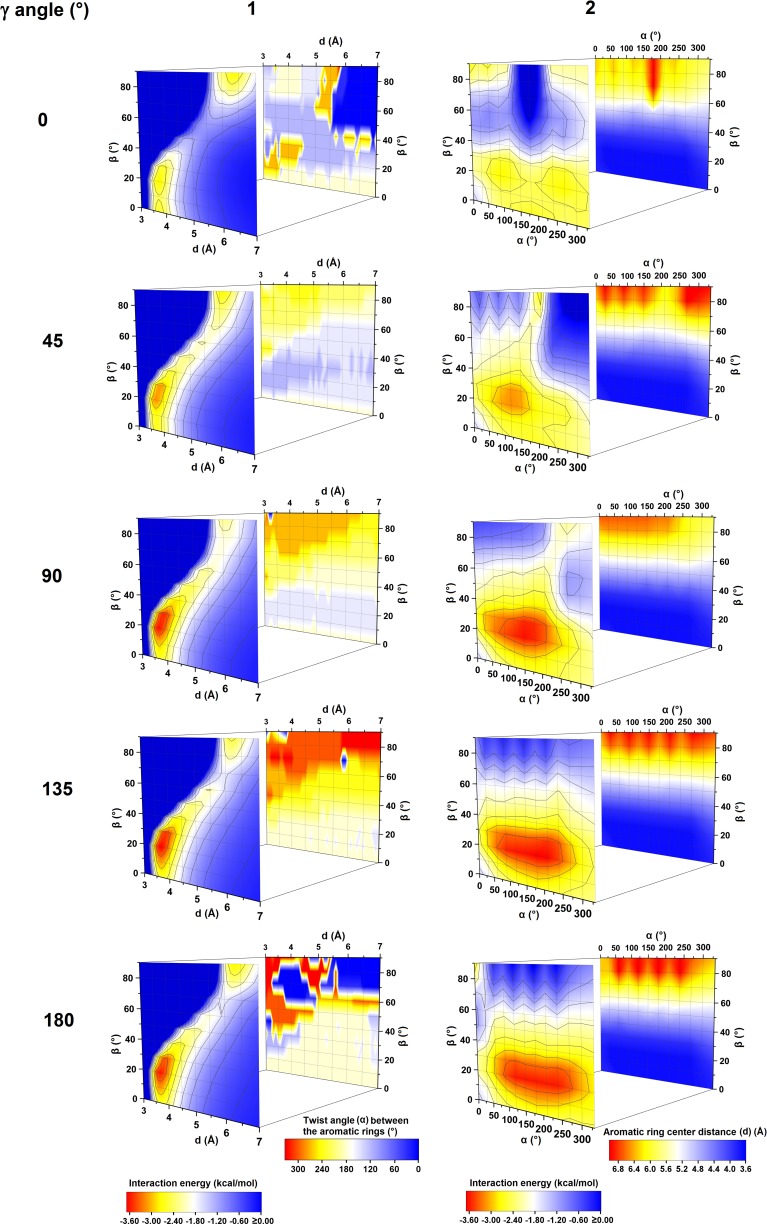
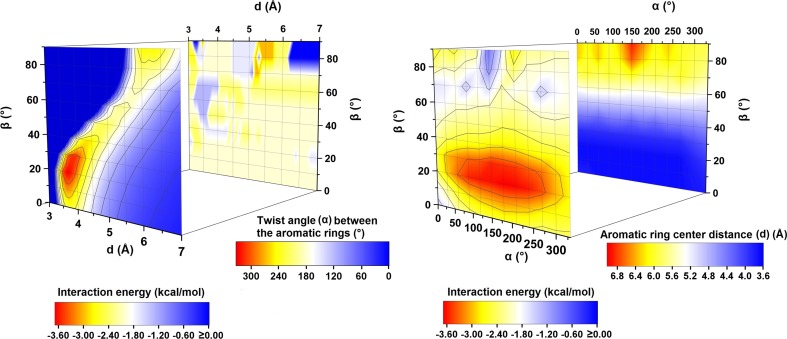
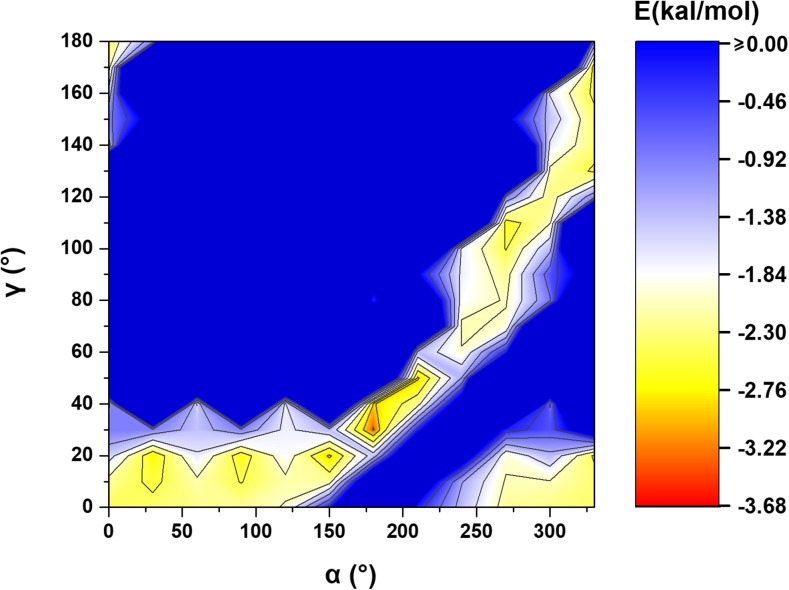
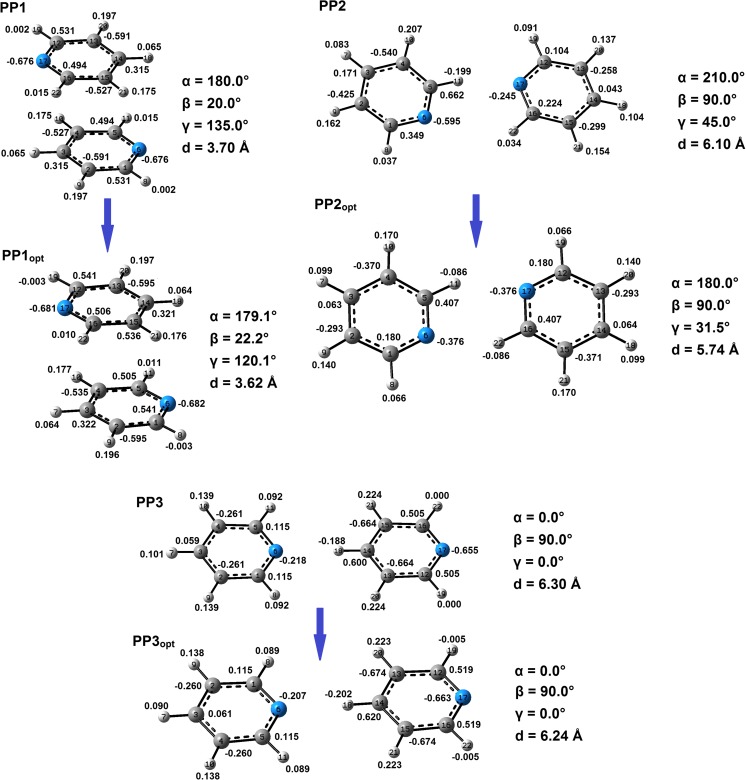
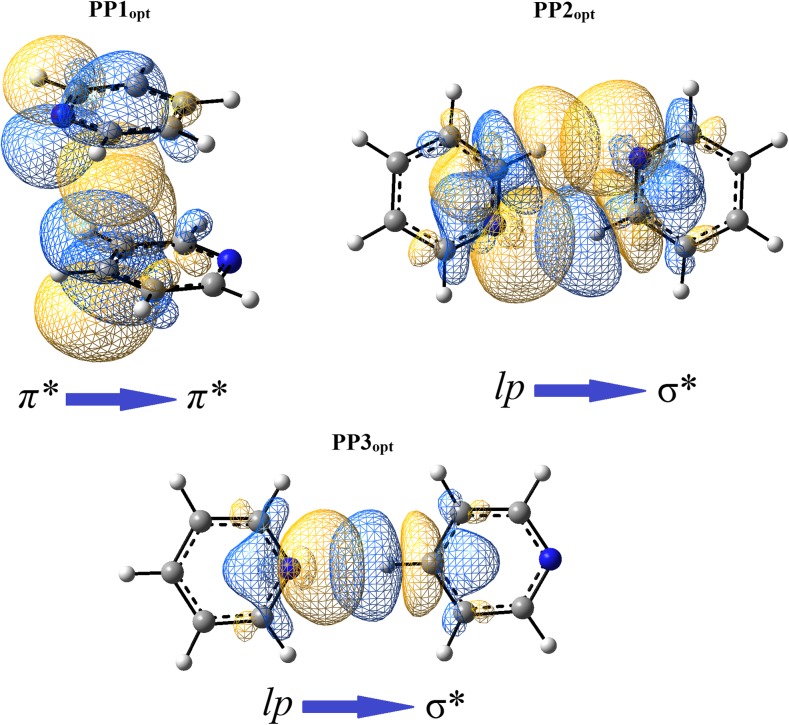
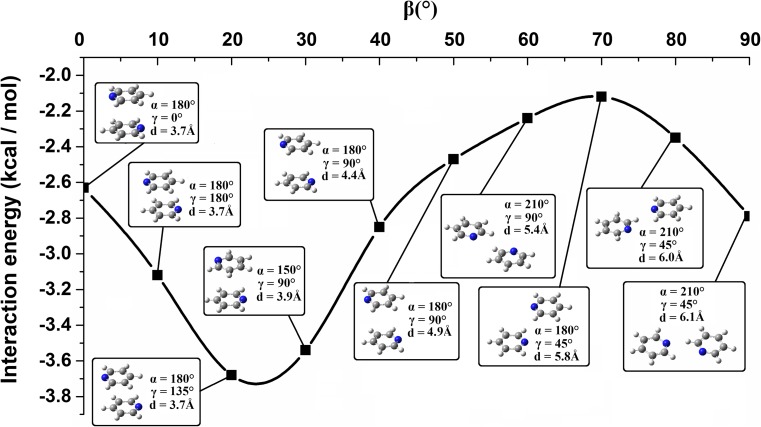
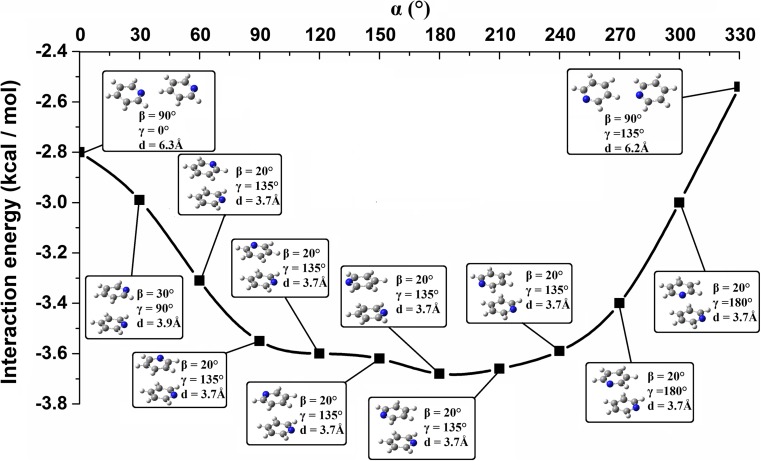
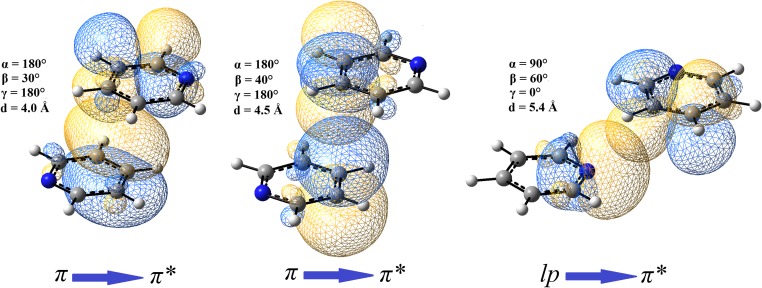
Extremely extensive calculations of potential energy surfaces for the parallel-displaced configuration of pyridine dimer systems have been carried out using a dispersion-corrected density functional. Instead of focusing on stationary geometries these calculations provide much deeper insight into the "landscape" of the interaction energies of the particular systems-one can learn how the pyridine dimer stability changes along with various geometrical parameters. Other calculations such as natural bond orbital and energy decomposition have also been applied. The interplay of two significant factors, electrostatic forces and electron correlation effects, have been evaluated. The role of π···π interactions in the stacked pyridine systems has also been confirmed, and surprisingly, this happened to be true even for the geometries where the formation of C-H···π interactions might be proposed instead. The combination of many different methods has revealed the complexity of the stacking interactions. Apart from providing a "literal new look" into pyridine interaction patterns another picture has emerged. A stacking interaction in a pyridine dimer system is perceived as a combination of many different sources of the interaction energy, including orbital ones, and this is true for many different geometries.
Keywords: Density-functional theory; Energy decomposition analysis; Natural bond orbital analysis; Pyridine; Stacking interactions.
Figures
Similar articles
-
Pi-pi interaction in pyridine.J Phys Chem A. 2005 Jan 13;109(1):6-8. doi: 10.1021/jp045218c. J Phys Chem A. 2005. PMID: 16839083
-
A new insight into π-π stacking involving remarkable orbital interactions.Phys Chem Chem Phys. 2016 Sep 14;18(36):25452-25457. doi: 10.1039/c6cp05485d. Phys Chem Chem Phys. 2016. PMID: 27711609
-
Orbital-based insights into parallel-displaced and twisted conformations in π-π interactions.Phys Chem Chem Phys. 2013 Jun 21;15(23):9397-406. doi: 10.1039/c3cp51077h. Epub 2013 May 10. Phys Chem Chem Phys. 2013. PMID: 23665910
-
Calculations on noncovalent interactions and databases of benchmark interaction energies.Acc Chem Res. 2012 Apr 17;45(4):663-72. doi: 10.1021/ar200255p. Epub 2012 Jan 6. Acc Chem Res. 2012. PMID: 22225511 Review.
-
Describing a landscape we are yet discovering.Adv Stat Anal. 2022;106(3):399-402. doi: 10.1007/s10182-022-00449-5. Epub 2022 Jun 9. Adv Stat Anal. 2022. PMID: 35698579 Free PMC article. Review. No abstract available.
Cited by
-
Tuning layered superstructures in precision polymers.Sci Rep. 2020 Jul 21;10(1):12119. doi: 10.1038/s41598-020-68927-x. Sci Rep. 2020. PMID: 32694581 Free PMC article.
-
Parallelized Raman Difference Spectroscopy for the Investigation of Chemical Interactions.Anal Chem. 2022 Jul 26;94(29):10346-10354. doi: 10.1021/acs.analchem.2c00222. Epub 2022 Jul 12. Anal Chem. 2022. PMID: 35820661 Free PMC article.
-
Interplay between Polymorphism and Isostructurality in the 2-Fur- and 2-Thenaldehyde Semi- and Thiosemicarbazones.Molecules. 2020 Feb 23;25(4):993. doi: 10.3390/molecules25040993. Molecules. 2020. PMID: 32102204 Free PMC article.
-
Excitation of rhodamine 800 in aqueous media: a theoretical investigation.J Mol Model. 2022 Feb 2;28(2):52. doi: 10.1007/s00894-022-05034-w. J Mol Model. 2022. PMID: 35112197
-
Theoretical calculations about nitro-substituted pyridine as high-energy-density compounds (HEDCs).J Mol Model. 2019 Jan 5;25(1):23. doi: 10.1007/s00894-018-3904-4. J Mol Model. 2019. PMID: 30612299
References
-
- Tsuzuki S. Interactions with aromatic rings. Struct Bond. 2005;115:149–193. doi: 10.1007/b135618. - DOI
-
- Wan CQ, Chen XD, Mak TCW. Supramolecular frameworks assembled via intermolecular lone pair-aromatic interaction between carbonyl and pyridyl groups. CrystEngComm. 2008;10:475–478. doi: 10.1039/b718823d. - DOI
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
