

Emerging functions of the EGFR in cancer
- PMID: 29124875
- PMCID: PMC5748484
- DOI: 10.1002/1878-0261.12155
Emerging functions of the EGFR in cancer
Abstract
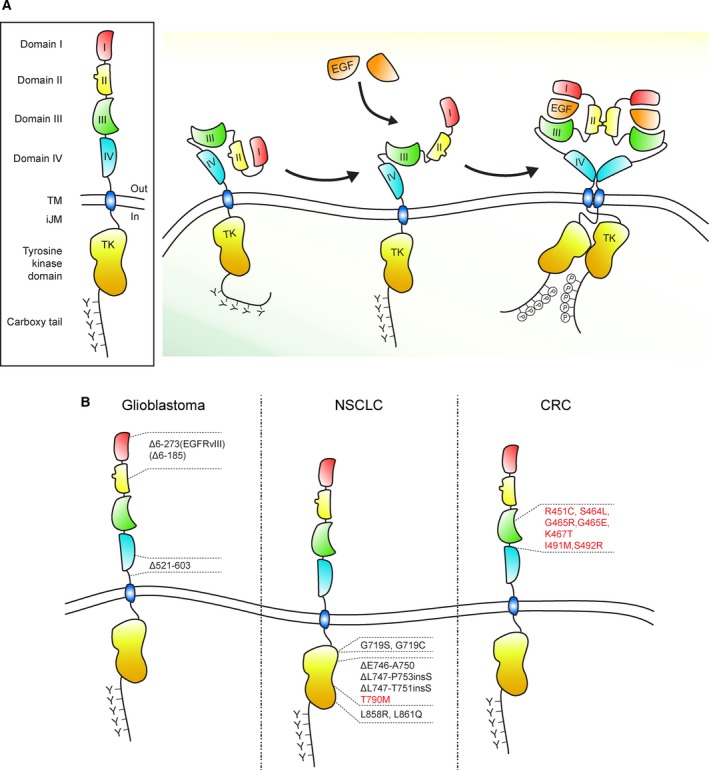
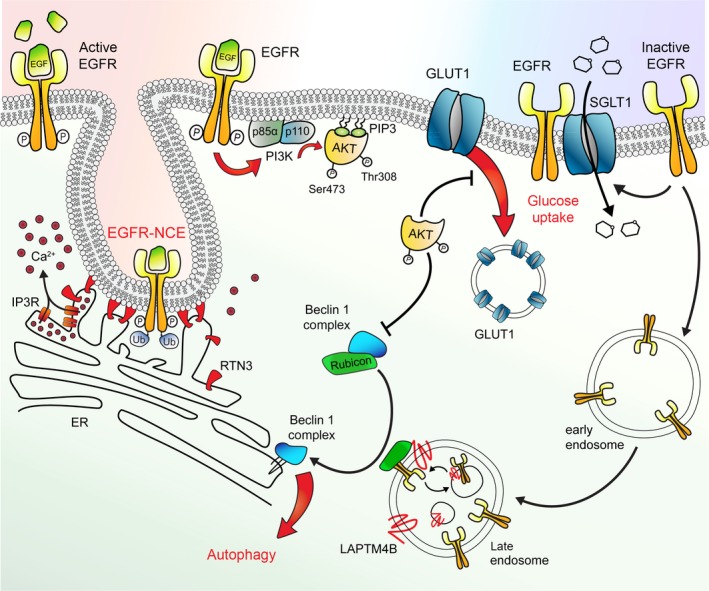
The physiological function of the epidermal growth factor receptor (EGFR) is to regulate epithelial tissue development and homeostasis. In pathological settings, mostly in lung and breast cancer and in glioblastoma, the EGFR is a driver of tumorigenesis. Inappropriate activation of the EGFR in cancer mainly results from amplification and point mutations at the genomic locus, but transcriptional upregulation or ligand overproduction due to autocrine/paracrine mechanisms has also been described. Moreover, the EGFR is increasingly recognized as a biomarker of resistance in tumors, as its amplification or secondary mutations have been found to arise under drug pressure. This evidence, in addition to the prominent function that this receptor plays in normal epithelia, has prompted intense investigations into the role of the EGFR both at physiological and at pathological level. Despite the large body of knowledge obtained over the last two decades, previously unrecognized (herein defined as 'noncanonical') functions of the EGFR are currently emerging. Here, we will initially review the canonical ligand-induced EGFR signaling pathway, with particular emphasis to its regulation by endocytosis and subversion in human tumors. We will then focus on the most recent advances in uncovering noncanonical EGFR functions in stress-induced trafficking, autophagy, and energy metabolism, with a perspective on future therapeutic applications.
Keywords: EGFR; cancer; membrane trafficking; signal transduction.
© 2017 The Authors. Published by FEBS Press and John Wiley & Sons Ltd.
Figures
References
-
- Arena S, Bellosillo B, Siravegna G, Martínez A, Cañadas I, Lazzari L, Ferruz N, Russo M, Misale S, González I et al (2015) Emergence of multiple EGFR extracellular mutations during cetuximab treatment in colorectal cancer. Clin Cancer Res 21, 2157–2166. - PubMed
-
- Arena S, Siravegna G, Mussolin B, Kearns JD, Wolf BB, Misale S, Lazzari L, Bertotti A, Trusolino L, Adjei AA et al (2016) MM‐151 overcomes acquired resistance to cetuximab and panitumumab in colorectal cancers harboring EGFR extracellular domain mutations. Sci Transl Med 8, 324ra314. - PubMed
-
- Arteaga CL, Ramsey TT, Shawver LK and Guyer CA (1997) Unliganded epidermal growth factor receptor dimerization induced by direct interaction of quinazolines with the ATP binding site. J Biol Chem 272, 23247–23254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
