

Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update
- PMID: 29125546
- PMCID: PMC5713354
- DOI: 10.3390/ijms18112385
Hereditary Nephrogenic Diabetes Insipidus: Pathophysiology and Possible Treatment. An Update
Abstract
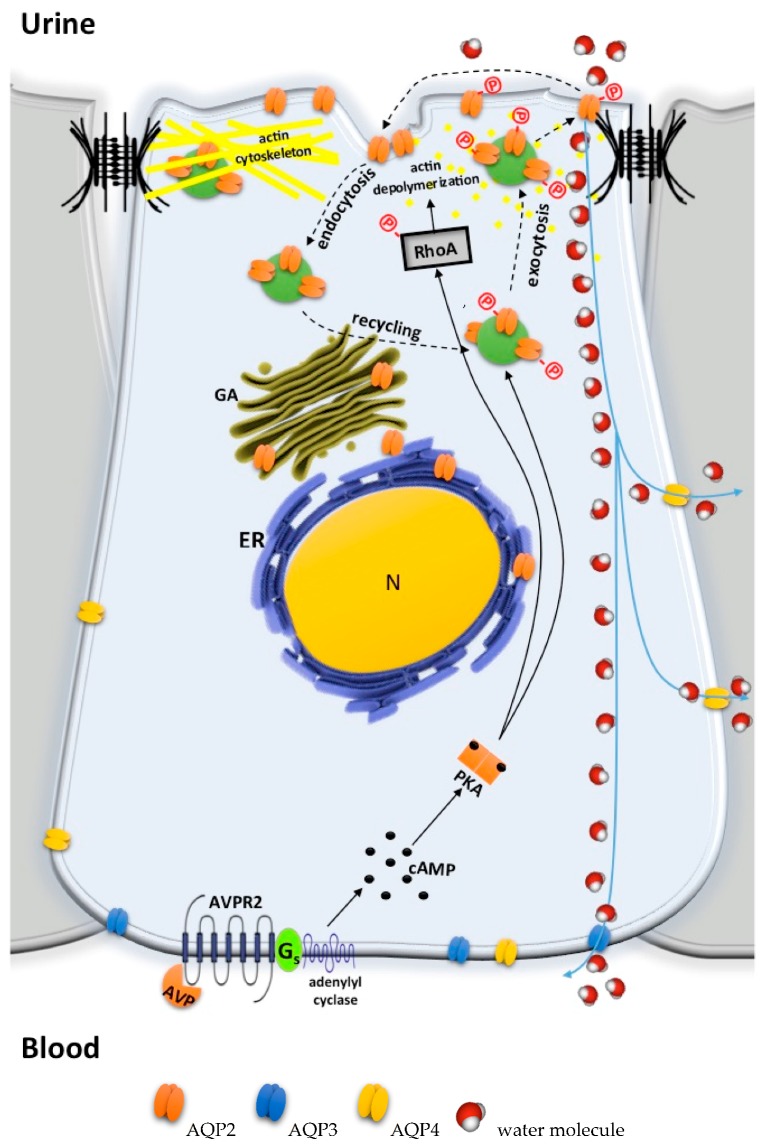
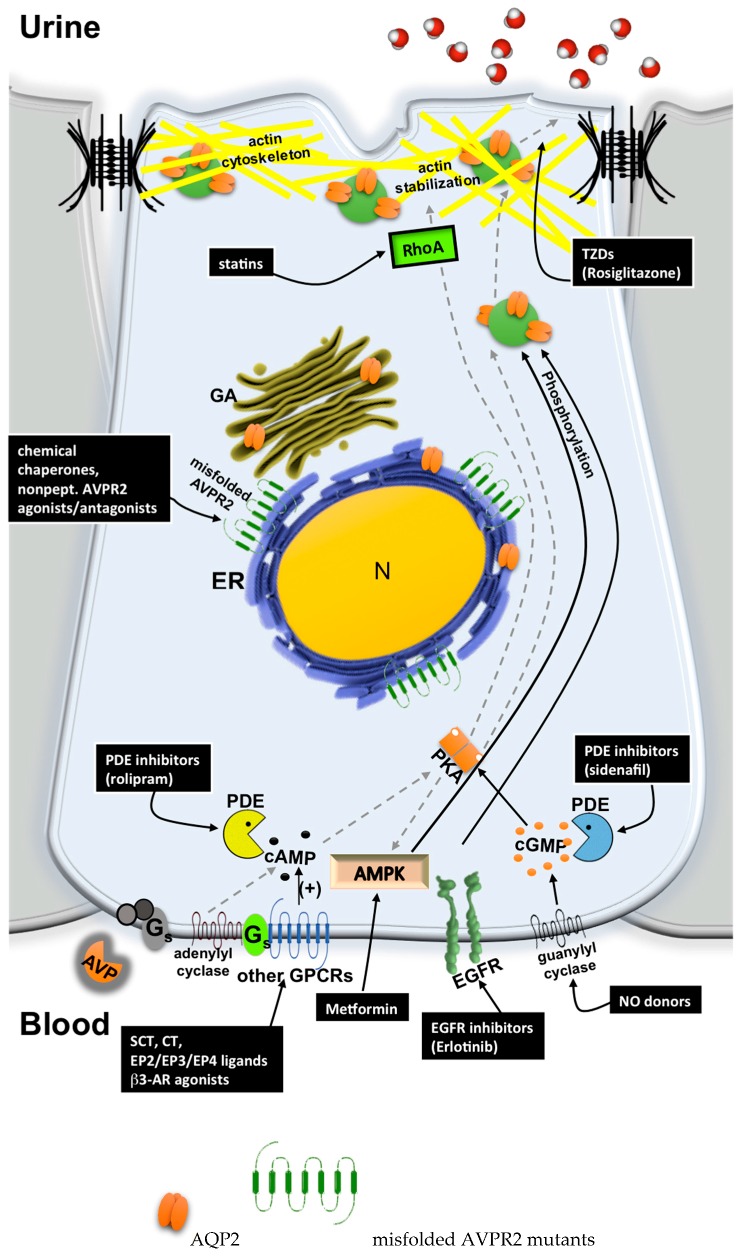
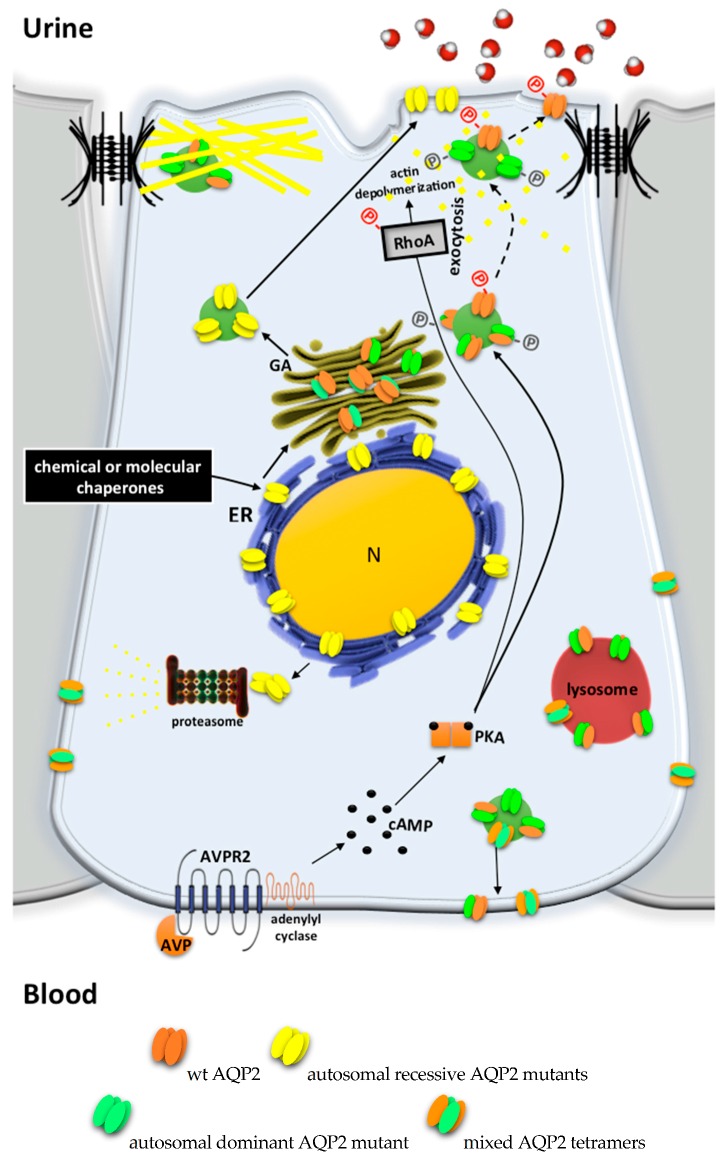
Under physiological conditions, excessive loss of water through the urine is prevented by the release of the antidiuretic hormone arginine-vasopressin (AVP) from the posterior pituitary. In the kidney, AVP elicits a number of cellular responses, which converge on increasing the osmotic reabsorption of water in the collecting duct. One of the key events triggered by the binding of AVP to its type-2 receptor (AVPR2) is the exocytosis of the water channel aquaporin 2 (AQP2) at the apical membrane the principal cells of the collecting duct. Mutations of either AVPR2 or AQP2 result in a genetic disease known as nephrogenic diabetes insipidus, which is characterized by the lack of responsiveness of the collecting duct to the antidiuretic action of AVP. The affected subject, being incapable of concentrating the urine, presents marked polyuria and compensatory polydipsia and is constantly at risk of severe dehydration. The molecular bases of the disease are fully uncovered, as well as the genetic or clinical tests for a prompt diagnosis of the disease in newborns. A real cure for nephrogenic diabetes insipidus (NDI) is still missing, and the main symptoms of the disease are handled with s continuous supply of water, a restrictive diet, and nonspecific drugs. Unfortunately, the current therapeutic options are limited and only partially beneficial. Further investigation in vitro or using the available animal models of the disease, combined with clinical trials, will eventually lead to the identification of one or more targeted strategies that will improve or replace the current conventional therapy and grant NDI patients a better quality of life. Here we provide an updated overview of the genetic defects causing NDI, the most recent strategies under investigation for rescuing the activity of mutated AVPR2 or AQP2, or for bypassing defective AVPR2 signaling and restoring AQP2 plasma membrane expression.
Keywords: antidiuresis; aquaporin-2 (AQP2); arginine-vasopressin (AVP); arginine-vasopressin receptor AVPR2; nephrogenic diabetes insipidus (NDI); polyuria.
Conflict of interest statement
The authors declare no conflicts of interest.
Figures
Similar articles
-
AQP2: Mutations Associated with Congenital Nephrogenic Diabetes Insipidus and Regulation by Post-Translational Modifications and Protein-Protein Interactions.Cells. 2020 Sep 26;9(10):2172. doi: 10.3390/cells9102172. Cells. 2020. PMID: 32993088 Free PMC article. Review.
-
Physiopathology and diagnosis of nephrogenic diabetes insipidus.Ann Endocrinol (Paris). 2012 Apr;73(2):128-9. doi: 10.1016/j.ando.2012.03.032. Epub 2012 Apr 13. Ann Endocrinol (Paris). 2012. PMID: 22503803 Review.
-
Genetic forms of nephrogenic diabetes insipidus (NDI): Vasopressin receptor defect (X-linked) and aquaporin defect (autosomal recessive and dominant).Best Pract Res Clin Endocrinol Metab. 2016 Mar;30(2):263-76. doi: 10.1016/j.beem.2016.02.010. Epub 2016 Mar 2. Best Pract Res Clin Endocrinol Metab. 2016. PMID: 27156763 Review.
-
Nephrogenic diabetes insipidus.Adv Chronic Kidney Dis. 2006 Apr;13(2):96-104. doi: 10.1053/j.ackd.2006.01.006. Adv Chronic Kidney Dis. 2006. PMID: 16580609 Review.
-
Nephrogenic diabetes insipidus: essential insights into the molecular background and potential therapies for treatment.Endocr Rev. 2013 Apr;34(2):278-301. doi: 10.1210/er.2012-1044. Epub 2013 Jan 29. Endocr Rev. 2013. PMID: 23360744 Free PMC article. Review.
Cited by
-
Treatment of Nephrogenic Diabetes Insipidus Patients With cGMP-Stimulating Drugs Does Not Mitigate Polyuria or Increase Urinary Concentrating Ability.Kidney Int Rep. 2020 May 23;5(8):1319-1325. doi: 10.1016/j.ekir.2020.05.015. eCollection 2020 Aug. Kidney Int Rep. 2020. PMID: 32775834 Free PMC article. No abstract available.
-
Vasopressin-Independent Regulation of Aquaporin-2 by Tamoxifen in Kidney Collecting Ducts.Front Physiol. 2019 Aug 9;10:948. doi: 10.3389/fphys.2019.00948. eCollection 2019. Front Physiol. 2019. PMID: 31447686 Free PMC article.
-
Renal water transport in health and disease.Pflugers Arch. 2022 Aug;474(8):841-852. doi: 10.1007/s00424-022-02712-9. Epub 2022 Jun 9. Pflugers Arch. 2022. PMID: 35678906 Free PMC article. Review.
-
Molecular characterization of an aquaporin-2 mutation causing a severe form of nephrogenic diabetes insipidus.Cell Mol Life Sci. 2020 Mar;77(5):953-962. doi: 10.1007/s00018-019-03219-w. Epub 2019 Jul 13. Cell Mol Life Sci. 2020. PMID: 31302751 Free PMC article.
-
Severe congenital nephrogenic diabetes insipidus in a compound heterozygote with a new large deletion of the AQP2 gene. A case report.Mol Genet Genomic Med. 2019 Apr;7(4):e00568. doi: 10.1002/mgg3.568. Epub 2019 Feb 19. Mol Genet Genomic Med. 2019. PMID: 30784238 Free PMC article.
References
-
- Robertson G.L. Physiology of adh secretion. Kidney Int. Suppl. 1987;21:S20–S26. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
