

Comprehensive analysis of gene expression patterns in Friedreich's ataxia fibroblasts by RNA sequencing reveals altered levels of protein synthesis factors and solute carriers
- PMID: 29125828
- PMCID: PMC5719256
- DOI: 10.1242/dmm.030536
Comprehensive analysis of gene expression patterns in Friedreich's ataxia fibroblasts by RNA sequencing reveals altered levels of protein synthesis factors and solute carriers
Abstract
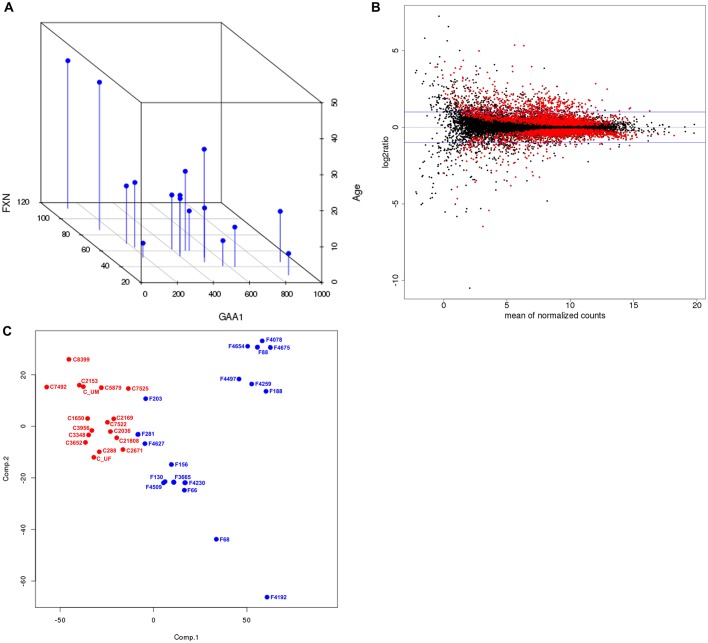
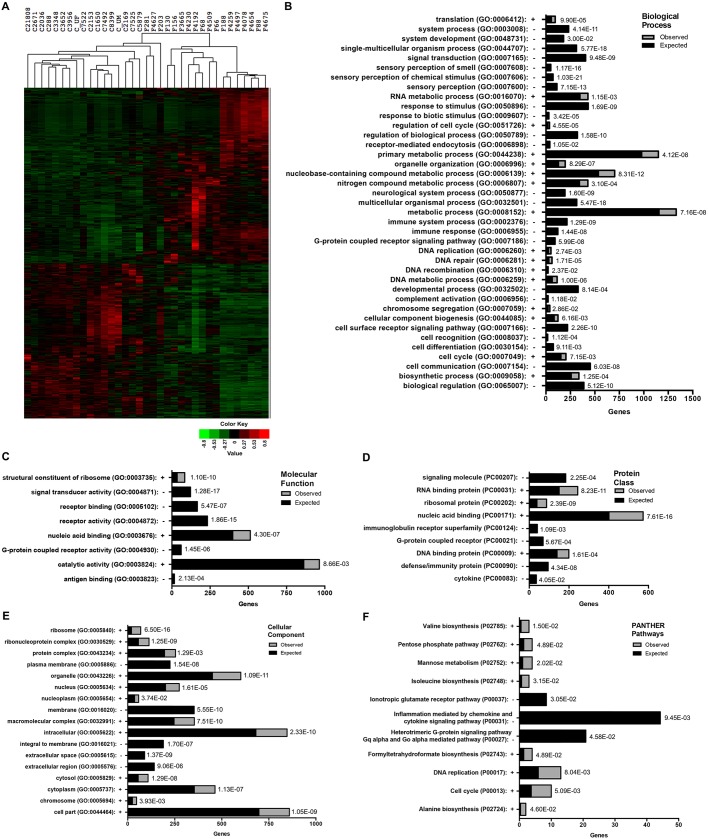
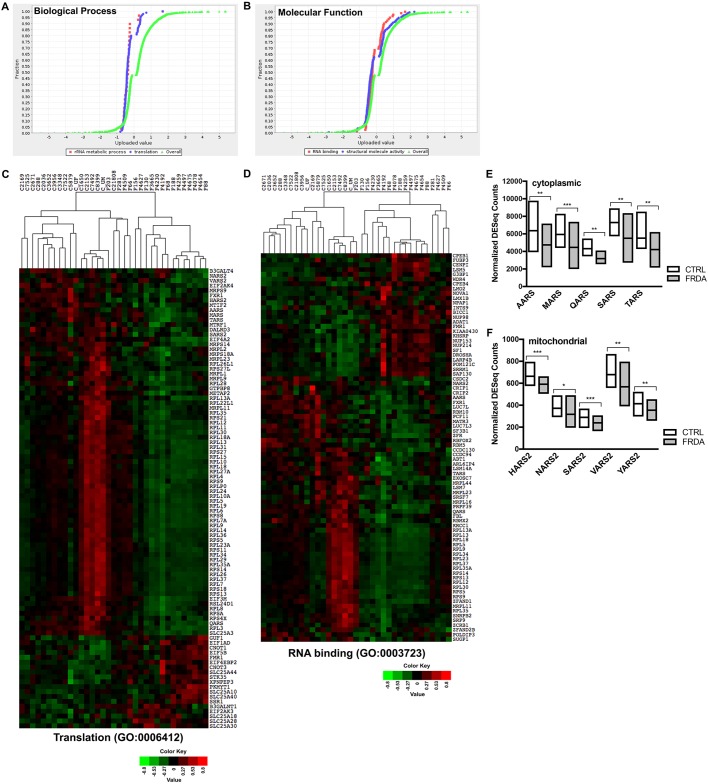
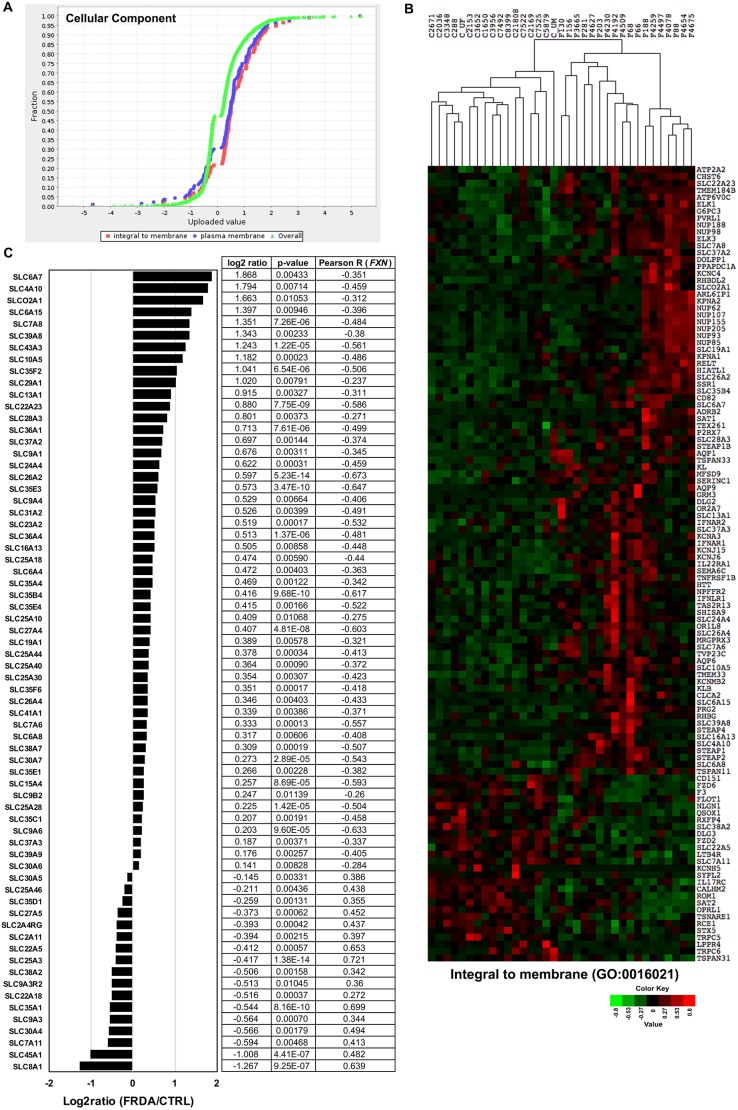
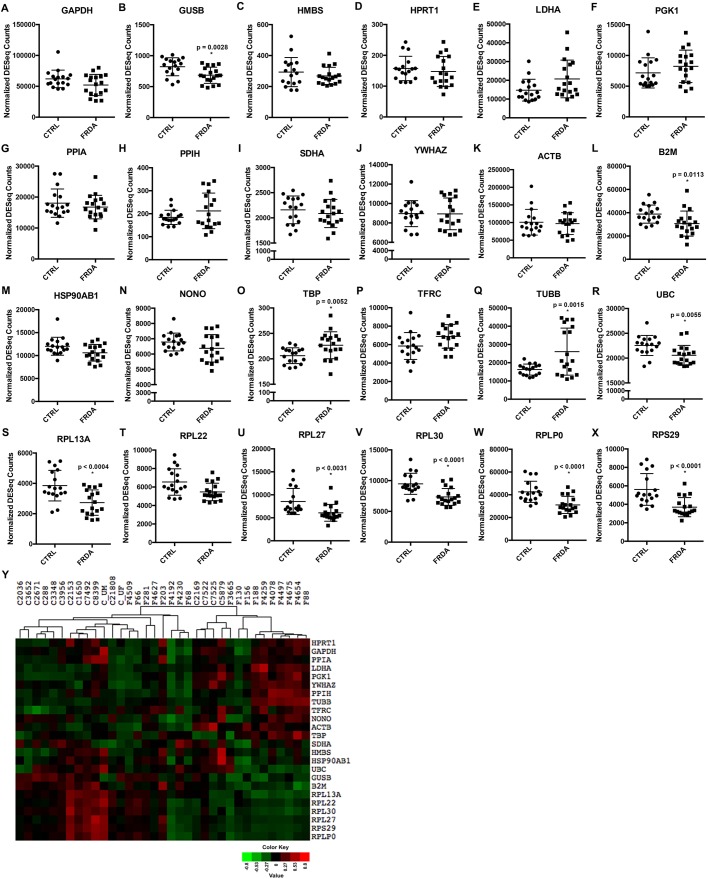
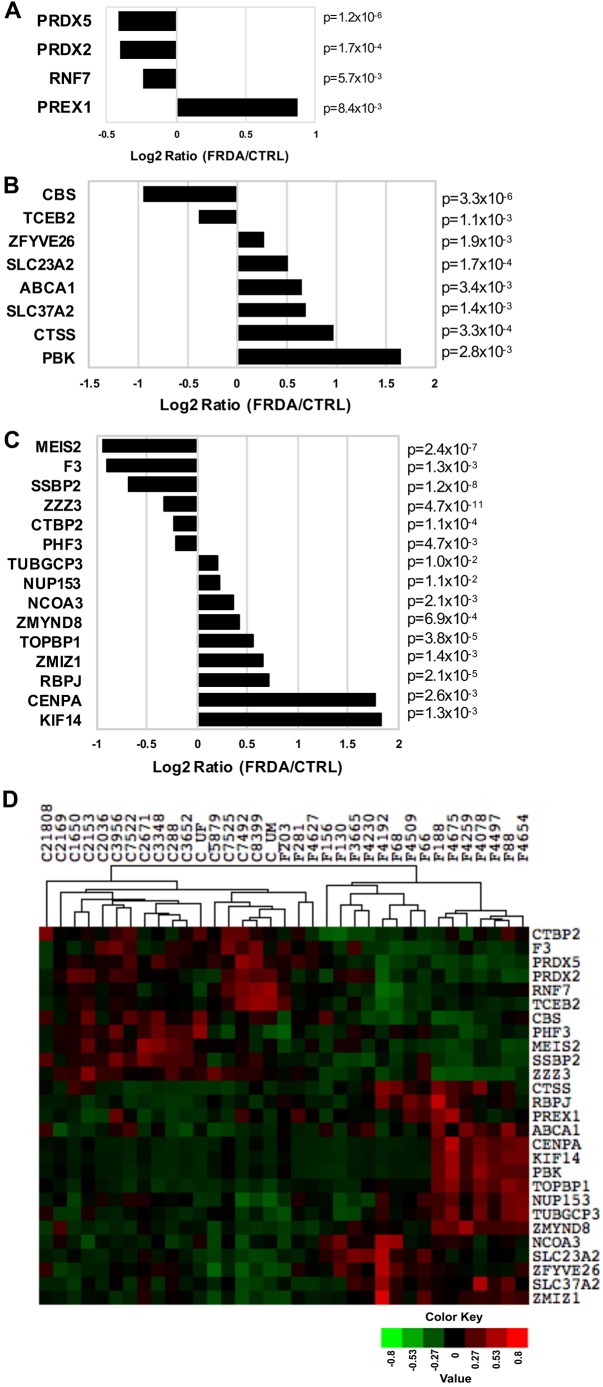
Friedreich's ataxia (FRDA) is an autosomal recessive neurodegenerative disease usually caused by large homozygous expansions of GAA repeat sequences in intron 1 of the frataxin (FXN) gene. FRDA patients homozygous for GAA expansions have low FXN mRNA and protein levels when compared with heterozygous carriers or healthy controls. Frataxin is a mitochondrial protein involved in iron-sulfur cluster synthesis, and many FRDA phenotypes result from deficiencies in cellular metabolism due to lowered expression of FXN Presently, there is no effective treatment for FRDA, and biomarkers to measure therapeutic trial outcomes and/or to gauge disease progression are lacking. Peripheral tissues, including blood cells, buccal cells and skin fibroblasts, can readily be isolated from FRDA patients and used to define molecular hallmarks of disease pathogenesis. For instance, FXN mRNA and protein levels as well as FXN GAA-repeat tract lengths are routinely determined using all of these cell types. However, because these tissues are not directly involved in disease pathogenesis, their relevance as models of the molecular aspects of the disease is yet to be decided. Herein, we conducted unbiased RNA sequencing to profile the transcriptomes of fibroblast cell lines derived from 18 FRDA patients and 17 unaffected control individuals. Bioinformatic analyses revealed significantly upregulated expression of genes encoding plasma membrane solute carrier proteins in FRDA fibroblasts. Conversely, the expression of genes encoding accessory factors and enzymes involved in cytoplasmic and mitochondrial protein synthesis was consistently decreased in FRDA fibroblasts. Finally, comparison of genes differentially expressed in FRDA fibroblasts to three previously published gene expression signatures defined for FRDA blood cells showed substantial overlap between the independent datasets, including correspondingly deficient expression of antioxidant defense genes. Together, these results indicate that gene expression profiling of cells derived from peripheral tissues can, in fact, consistently reveal novel molecular pathways of the disease. When performed on statistically meaningful sample group sizes, unbiased global profiling analyses utilizing peripheral tissues are critical for the discovery and validation of FRDA disease biomarkers.
Keywords: Fibroblasts; Friedreich's ataxia; RNA sequencing; Solute carriers; Translation.
© 2017. Published by The Company of Biologists Ltd.
Conflict of interest statement
Competing interestsThe authors declare no competing or financial interests.
Figures
Similar articles
-
A Comprehensive Transcriptome Analysis Identifies FXN and BDNF as Novel Targets of miRNAs in Friedreich's Ataxia Patients.Mol Neurobiol. 2020 Jun;57(6):2639-2653. doi: 10.1007/s12035-020-01899-1. Epub 2020 Apr 14. Mol Neurobiol. 2020. PMID: 32291635 Free PMC article.
-
Mitochondrial damage and senescence phenotype of cells derived from a novel frataxin G127V point mutation mouse model of Friedreich's ataxia.Dis Model Mech. 2020 Jul 27;13(7):dmm045229. doi: 10.1242/dmm.045229. Dis Model Mech. 2020. PMID: 32586831 Free PMC article.
-
Alleviating GAA Repeat Induced Transcriptional Silencing of the Friedreich's Ataxia Gene During Somatic Cell Reprogramming.Stem Cells Dev. 2016 Dec 1;25(23):1788-1800. doi: 10.1089/scd.2016.0147. Epub 2016 Oct 17. Stem Cells Dev. 2016. PMID: 27615158 Free PMC article.
-
Beyond loss of frataxin: the complex molecular pathology of Friedreich ataxia.Discov Med. 2014 Jan;17(91):25-35. Discov Med. 2014. PMID: 24411698 Review.
-
Friedreich's ataxia and frataxin: molecular genetics, evolution and pathogenesis (Review).Int J Mol Med. 2001 Jun;7(6):581-9. doi: 10.3892/ijmm.7.6.581. Int J Mol Med. 2001. PMID: 11351269 Review.
Cited by
-
G-rich motifs within phosphorothioate-based antisense oligonucleotides (ASOs) drive activation of FXN expression through indirect effects.Nucleic Acids Res. 2022 Dec 9;50(22):12657-12673. doi: 10.1093/nar/gkac1108. Nucleic Acids Res. 2022. PMID: 36511872 Free PMC article.
-
Targeting 3' and 5' untranslated regions with antisense oligonucleotides to stabilize frataxin mRNA and increase protein expression.Nucleic Acids Res. 2021 Nov 18;49(20):11560-11574. doi: 10.1093/nar/gkab954. Nucleic Acids Res. 2021. PMID: 34718736 Free PMC article.
-
The Multifaceted Roles of Proline in Cell Behavior.Front Cell Dev Biol. 2021 Aug 12;9:728576. doi: 10.3389/fcell.2021.728576. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34458276 Free PMC article. Review.
-
Frataxin controls ketone body metabolism through regulation of OXCT1.PNAS Nexus. 2022 Jul 26;1(3):pgac142. doi: 10.1093/pnasnexus/pgac142. eCollection 2022 Jul. PNAS Nexus. 2022. PMID: 36016708 Free PMC article.
-
Defining Transcription Regulatory Elements in the Human Frataxin Gene: Implications for Gene Therapy.Hum Gene Ther. 2020 Aug;31(15-16):839-851. doi: 10.1089/hum.2020.053. Epub 2020 Jul 13. Hum Gene Ther. 2020. PMID: 32527155 Free PMC article.
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
