

Effects of cognate, non-cognate and synthetic CXCR4 and ACKR3 ligands on human lung endothelial cell barrier function
- PMID: 29125867
- PMCID: PMC5681266
- DOI: 10.1371/journal.pone.0187949
Effects of cognate, non-cognate and synthetic CXCR4 and ACKR3 ligands on human lung endothelial cell barrier function
Abstract
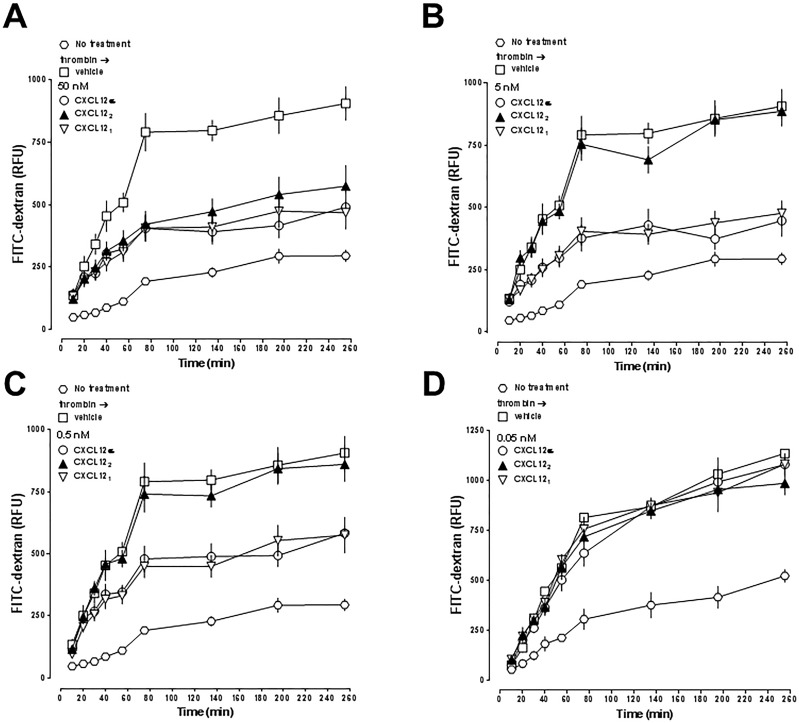
Recent evidence suggests that chemokine CXCL12, the cognate agonist of chemokine receptors CXCR4 and ACKR3, reduces thrombin-mediated impairment of endothelial barrier function. A detailed characterization of the effects of CXCL12 on thrombin-mediated human lung endothelial hyperpermeability is lacking and structure-function correlations are not available. Furthermore, effects of other CXCR4/ACKR3 ligands on lung endothelial barrier function are unknown. Thus, we tested the effects of a panel of CXCR4/ACKR3 ligands (CXCL12, CXCL11, ubiquitin, AMD3100, TC14012) and compared the CXCR4/ACKR3 activities of CXCL12 variants (CXCL12α/β, CXCL12(3-68), CXCL121, CXCL122, CXCL12-S-S4V, CXCL12-R47E, CXCL12-K27A/R41A/R47A) with their effects on human lung endothelial barrier function in permeability assays. CXCL12α enhanced human primary pulmonary artery endothelial cell (hPPAEC) barrier function, whereas CXCL11, ubiquitin, AMD3100 and TC14012 were ineffective. Pre-treatment of hPPAEC with CXCL12α and ubiquitin reduced thrombin-mediated hyperpermeability. CXCL12α-treatment of hPPAEC after thrombin exposure reduced barrier function impairment by 70% (EC50 0.05-0.5nM), which could be antagonized with AMD3100; ubiquitin (0.03-3μM) was ineffective. In a human lung microvascular endothelial cell line (HULEC5a), CXCL12α and ubiquitin post-treatment attenuated thrombin-induced hyperpermeability to a similar degree. CXCL12(3-68) was inefficient to activate CXCR4 in Presto-Tango β-arrestin2 recruitment assays; CXCL12-S-S4V, CXCL12-R47E and CXCL12-K27A/R41A/R47A showed significantly reduced potencies to activate CXCR4. While the potencies of all proteins in ACKR3 Presto-Tango assays were comparable, the efficacy of CXCL12(3-68) to activate ACKR3 was significantly reduced. The potencies to attenuate thrombin-mediated hPPAEC barrier function impairment were: CXCL12α/β, CXCL121, CXCL12-K27A/R41A/R47A > CXCL12-S-S4V, CXCL12-R47E > CXCL122 > CXCL12(3-68). Our findings indicate that CXCR4 activation attenuates thrombin-induced lung endothelial barrier function impairment and suggest that protective effects of CXCL12 are dictated by its CXCR4 agonist activity and interactions of distinct protein moieties with heparan sulfate on the endothelial surface. These data may facilitate development of compounds with improved pharmacological properties to attenuate thrombin-induced vascular leakage in the pulmonary circulation.
Conflict of interest statement
Figures








Similar articles
-
Chemokine (C-X-C motif) receptor 4 and atypical chemokine receptor 3 regulate vascular α₁-adrenergic receptor function.Mol Med. 2014 Oct 13;20(1):435-47. doi: 10.2119/molmed.2014.00101. Mol Med. 2014. PMID: 25032954 Free PMC article.
-
Partial agonist activity of α1-adrenergic receptor antagonists for chemokine (C-X-C motif) receptor 4 and atypical chemokine receptor 3.PLoS One. 2018 Sep 24;13(9):e0204041. doi: 10.1371/journal.pone.0204041. eCollection 2018. PLoS One. 2018. PMID: 30248140 Free PMC article.
-
Differential activity and selectivity of N-terminal modified CXCL12 chemokines at the CXCR4 and ACKR3 receptors.J Leukoc Biol. 2020 Jun;107(6):1123-1135. doi: 10.1002/JLB.2MA0320-383RR. Epub 2020 May 6. J Leukoc Biol. 2020. PMID: 32374043 Free PMC article.
-
The relevance of the chemokine receptor ACKR3/CXCR7 on CXCL12-mediated effects in cancers with a focus on virus-related cancers.Cytokine Growth Factor Rev. 2014 Jun;25(3):307-16. doi: 10.1016/j.cytogfr.2014.04.006. Epub 2014 May 9. Cytokine Growth Factor Rev. 2014. PMID: 24853339 Review.
-
Crosstalk between CXCL12/CXCR4/ACKR3 and the STAT3 Pathway.Cells. 2024 Jun 13;13(12):1027. doi: 10.3390/cells13121027. Cells. 2024. PMID: 38920657 Free PMC article. Review.
Cited by
-
Class A G protein-coupled receptors assemble into functional higher-order hetero-oligomers.FEBS Lett. 2021 Jul;595(14):1863-1875. doi: 10.1002/1873-3468.14135. Epub 2021 Jun 11. FEBS Lett. 2021. PMID: 34032285 Free PMC article.
-
Identifying and Assessing Putative Allosteric Sites and Modulators for CXCR4 Predicted through Network Modeling and Site Identification by Ligand Competitive Saturation.J Phys Chem B. 2024 May 30;128(21):5157-5174. doi: 10.1021/acs.jpcb.4c00925. Epub 2024 Apr 22. J Phys Chem B. 2024. PMID: 38647430 Free PMC article.
-
Natural and engineered chemokine (C-X-C motif) receptor 4 agonists prevent acute respiratory distress syndrome after lung ischemia-reperfusion injury and hemorrhage.Sci Rep. 2020 Jul 9;10(1):11359. doi: 10.1038/s41598-020-68425-0. Sci Rep. 2020. PMID: 32647374 Free PMC article.
-
Beta-Arrestins and Receptor Signaling in the Vascular Endothelium.Biomolecules. 2020 Dec 23;11(1):9. doi: 10.3390/biom11010009. Biomolecules. 2020. PMID: 33374806 Free PMC article. Review.
-
Stromal cell-derived factor-1 (CXCL12) and its role in bone and muscle biology.Cytokine. 2019 Nov;123:154783. doi: 10.1016/j.cyto.2019.154783. Epub 2019 Jul 20. Cytokine. 2019. PMID: 31336263 Free PMC article. Review.
References
-
- Blank R, Napolitano LM. Epidemiology of ARDS and ALI. Crit Care Clin. 2011;27(3):439–58. doi: 10.1016/j.ccc.2011.05.005 . - DOI - PubMed
-
- Ranieri VM, Rubenfeld GD, Thompson BT, Ferguson ND, Caldwell E, Fan E, et al. Acute respiratory distress syndrome: the Berlin Definition. JAMA. 2012;307(23):2526–33. Epub 2012/07/17. doi: 10.1001/jama.2012.5669 . - DOI - PubMed
-
- Dushianthan A, Grocott MP, Postle AD, Cusack R. Acute respiratory distress syndrome and acute lung injury. Postgrad Med J. 2011;87(1031):612–22. doi: 10.1136/pgmj.2011.118398 . - DOI - PubMed
-
- Ware LB. Pathophysiology of acute lung injury and the acute respiratory distress syndrome. Semin Respir Crit Care Med. 2006;27(4):337–49. Epub 2006/08/16. doi: 10.1055/s-2006-948288 . - DOI - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
