

Molecular insights into cortico-striatal miscommunications in Huntington's disease
- PMID: 29125980
- PMCID: PMC5825262
- DOI: 10.1016/j.conb.2017.10.019
Molecular insights into cortico-striatal miscommunications in Huntington's disease
Abstract
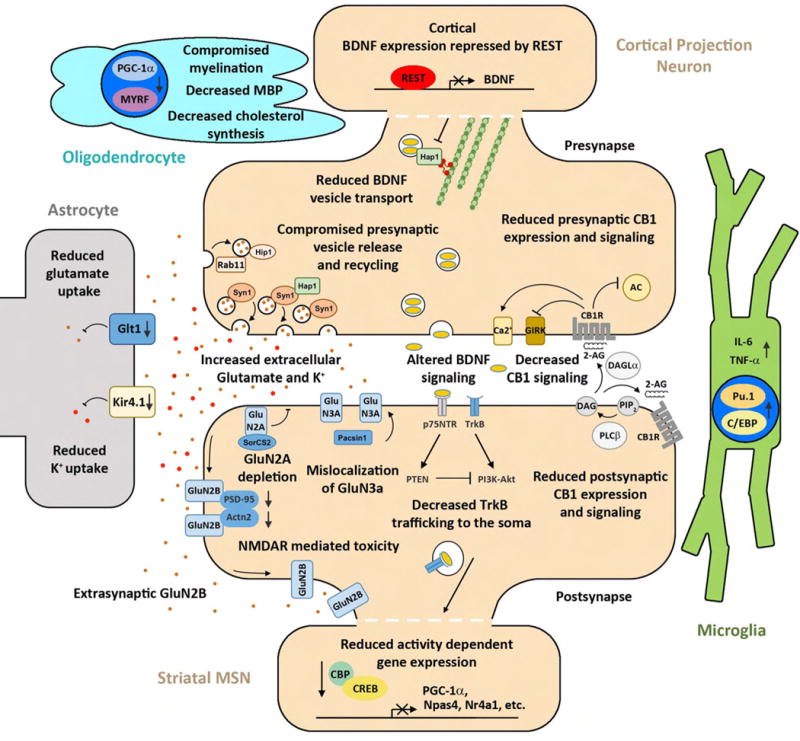
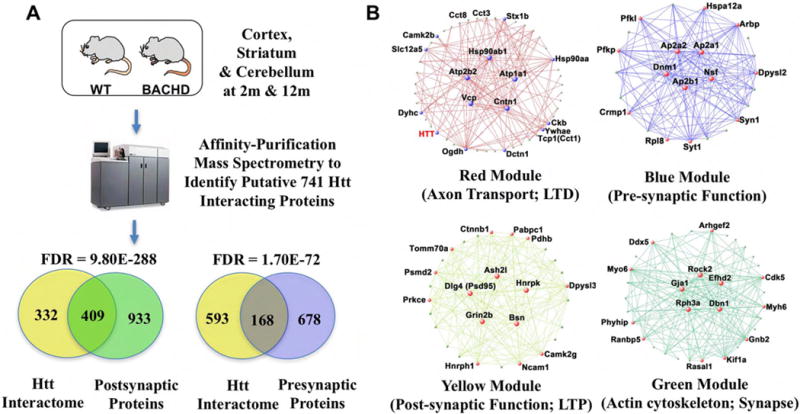
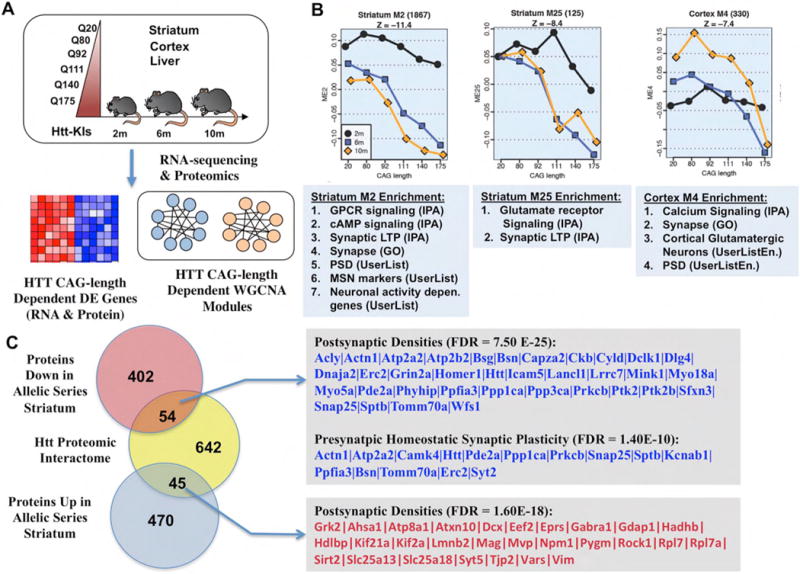
Huntington's disease (HD), a dominantly inherited neurodegenerative disease, is defined by its genetic cause, a CAG-repeat expansion in the HTT gene, its motor and psychiatric symptomology and primary loss of striatal medium spiny neurons (MSNs). However, the molecular mechanisms from genetic lesion to disease phenotype remain largely unclear. Mouse models of HD have been created that exhibit phenotypes partially recapitulating those in the patient, and specifically, cortico-striatal disconnectivity appears to be a shared pathogenic event shared by HD mouse models and patients. Molecular studies have begun to unveil converging molecular and cellular pathogenic mechanisms that may account for cortico-striatal miscommunication in various HD mouse models. Systems biological approaches help to illuminate synaptic molecular networks as a nexus for HD cortio-striatal pathogenesis, and may offer new candidate targets to modify the disease.
Copyright © 2017 Elsevier Ltd. All rights reserved.
Figures

References
-
- Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, Scahill RI, Leavitt BR, Stout JC, Paulsen JS, et al. Huntington disease: natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–216. - PubMed
-
- Saudou F, Humbert S. The Biology of Huntingtin. Neuron. 2016;89:910–926. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
