

Hereditary spastic paraplegia type 5: natural history, biomarkers and a randomized controlled trial
- PMID: 29126212
- PMCID: PMC5841036
- DOI: 10.1093/brain/awx273
Hereditary spastic paraplegia type 5: natural history, biomarkers and a randomized controlled trial
Abstract
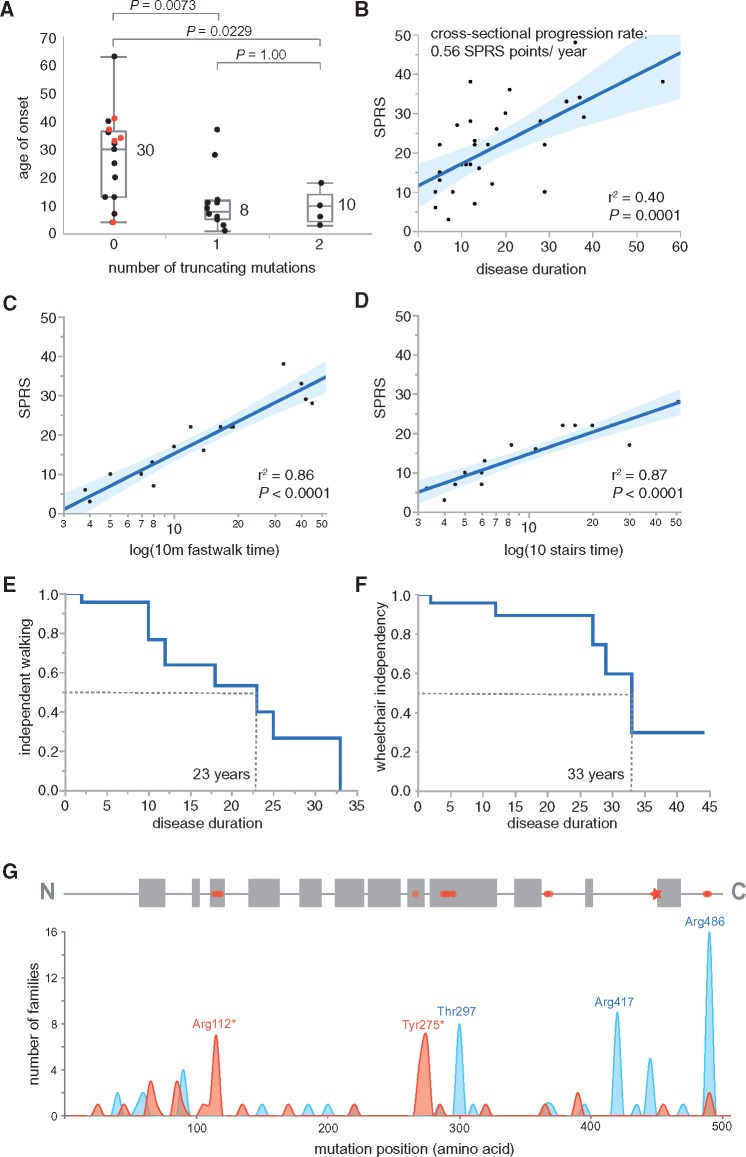
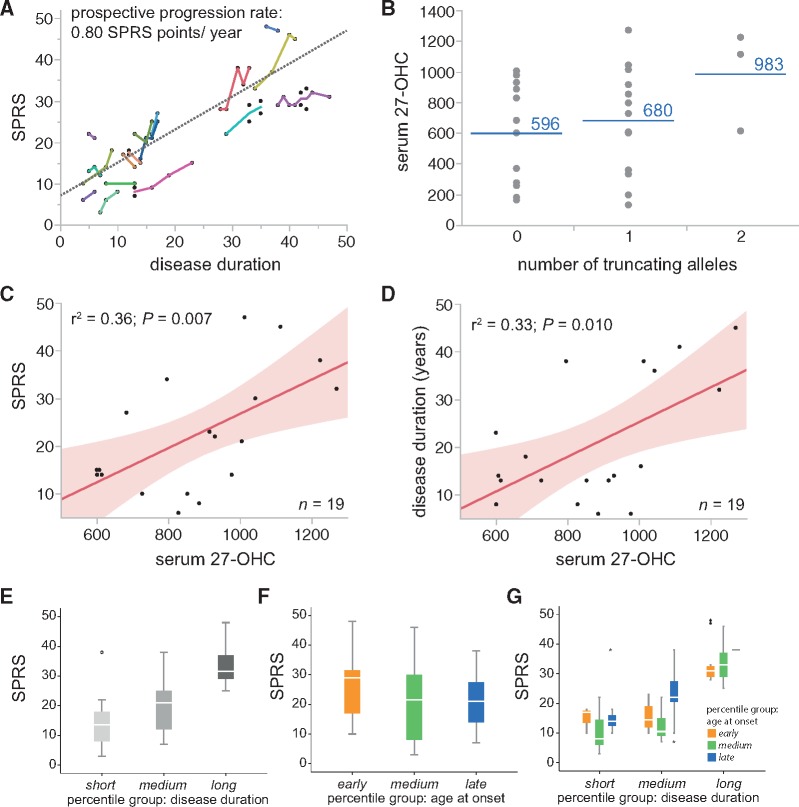
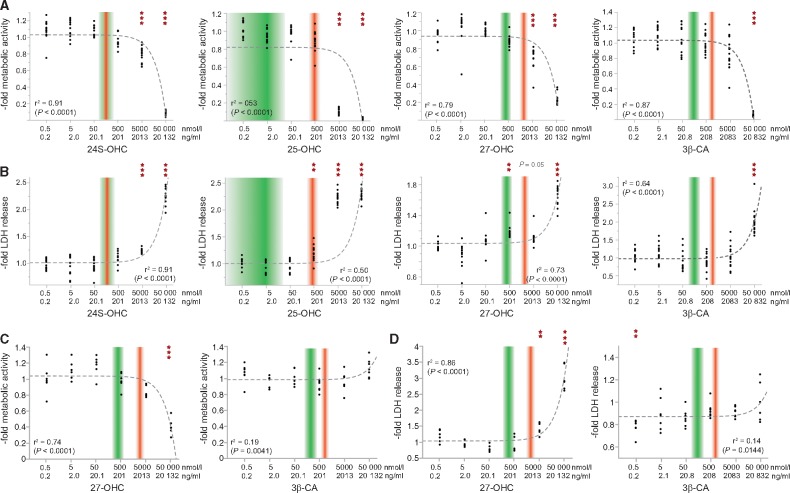
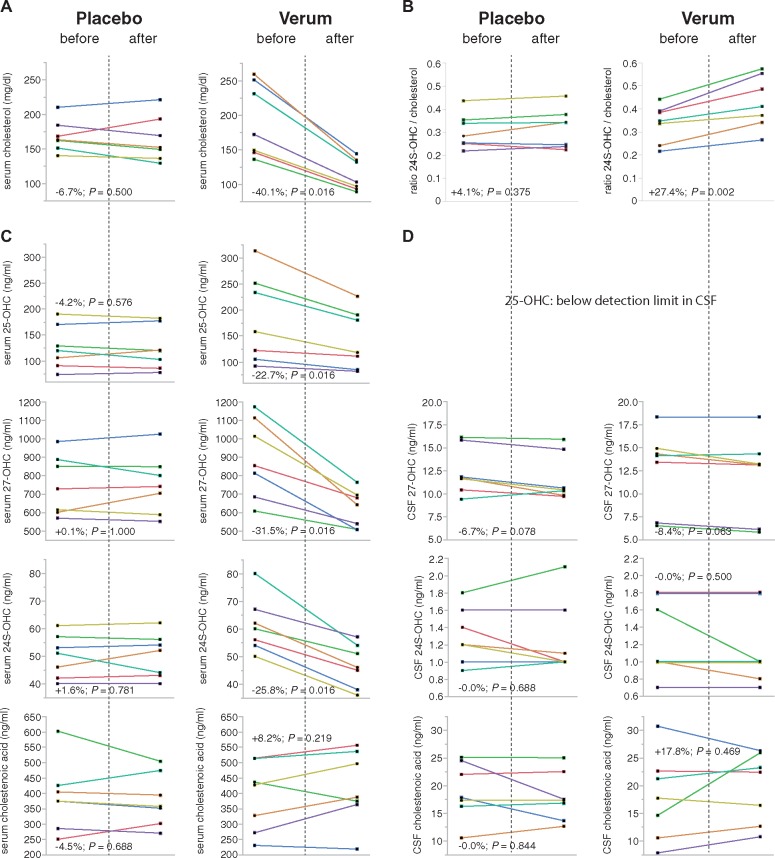
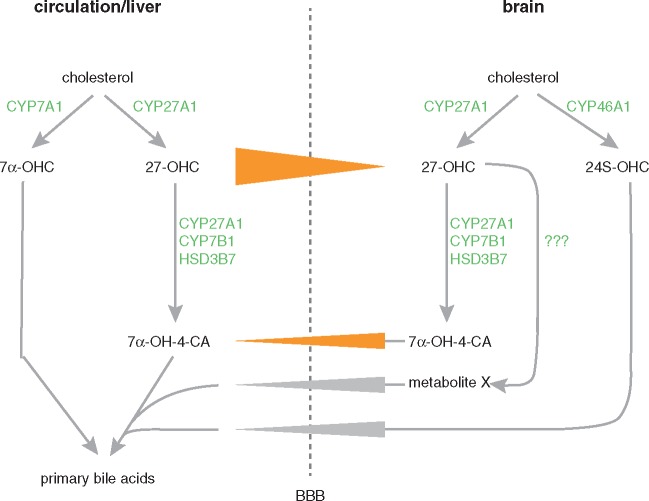
Spastic paraplegia type 5 (SPG5) is a rare subtype of hereditary spastic paraplegia, a highly heterogeneous group of neurodegenerative disorders defined by progressive neurodegeneration of the corticospinal tract motor neurons. SPG5 is caused by recessive mutations in the gene CYP7B1 encoding oxysterol-7α-hydroxylase. This enzyme is involved in the degradation of cholesterol into primary bile acids. CYP7B1 deficiency has been shown to lead to accumulation of neurotoxic oxysterols. In this multicentre study, we have performed detailed clinical and biochemical analysis in 34 genetically confirmed SPG5 cases from 28 families, studied dose-dependent neurotoxicity of oxysterols in human cortical neurons and performed a randomized placebo-controlled double blind interventional trial targeting oxysterol accumulation in serum of SPG5 patients. Clinically, SPG5 manifested in childhood or adolescence (median 13 years). Gait ataxia was a common feature. SPG5 patients lost the ability to walk independently after a median disease duration of 23 years and became wheelchair dependent after a median 33 years. The overall cross-sectional progression rate of 0.56 points on the Spastic Paraplegia Rating Scale per year was slightly lower than the longitudinal progression rate of 0.80 points per year. Biochemically, marked accumulation of CYP7B1 substrates including 27-hydroxycholesterol was confirmed in serum (n = 19) and cerebrospinal fluid (n = 17) of SPG5 patients. Moreover, 27-hydroxycholesterol levels in serum correlated with disease severity and disease duration. Oxysterols were found to impair metabolic activity and viability of human cortical neurons at concentrations found in SPG5 patients, indicating that elevated levels of oxysterols might be key pathogenic factors in SPG5. We thus performed a randomized placebo-controlled trial (EudraCT 2015-000978-35) with atorvastatin 40 mg/day for 9 weeks in 14 SPG5 patients with 27-hydroxycholesterol levels in serum as the primary outcome measure. Atorvastatin, but not placebo, reduced serum 27-hydroxycholesterol from 853 ng/ml [interquartile range (IQR) 683-1113] to 641 (IQR 507-694) (-31.5%, P = 0.001, Mann-Whitney U-test). Similarly, 25-hydroxycholesterol levels in serum were reduced. In cerebrospinal fluid 27-hydroxycholesterol was reduced by 8.4% but this did not significantly differ from placebo. As expected, no effects were seen on clinical outcome parameters in this short-term trial. In this study, we define the mutational and phenotypic spectrum of SPG5, examine the correlation of disease severity and progression with oxysterol concentrations, and demonstrate in a randomized controlled trial that atorvastatin treatment can effectively lower 27-hydroxycholesterol levels in serum of SPG5 patients. We thus demonstrate the first causal treatment strategy in hereditary spastic paraplegia.
Keywords: SPG5; biomarker; hereditary spastic paraplegia; oxysterol; randomized controlled trial.
© The Author (2017). Published by Oxford University Press on behalf of the Guarantors of Brain. All rights reserved. For Permissions, please email: journals.permissions@oup.com.
Figures
Similar articles
-
Plasma oxysterols: biomarkers for diagnosis and treatment in spastic paraplegia type 5.Brain. 2018 Jan 1;141(1):72-84. doi: 10.1093/brain/awx297. Brain. 2018. PMID: 29228183 Clinical Trial.
-
Mining for Oxysterols in Cyp7b1-/- Mouse Brain and Plasma: Relevance to Spastic Paraplegia Type 5.Biomolecules. 2019 Apr 13;9(4):149. doi: 10.3390/biom9040149. Biomolecules. 2019. PMID: 31013940 Free PMC article.
-
Elevated hydroxycholesterols in Norwegian patients with hereditary spastic paraplegia SPG5.J Neurol Sci. 2020 Dec 15;419:117211. doi: 10.1016/j.jns.2020.117211. Epub 2020 Oct 29. J Neurol Sci. 2020. PMID: 33160247
-
Childhood-onset hereditary spastic paraplegia and its treatable mimics.Mol Genet Metab. 2022 Dec;137(4):436-444. doi: 10.1016/j.ymgme.2021.06.006. Epub 2021 Jun 24. Mol Genet Metab. 2022. PMID: 34183250 Free PMC article. Review.
-
On the fluxes of side-chain oxidized oxysterols across blood-brain and blood-CSF barriers and origin of these steroids in CSF (Review).J Steroid Biochem Mol Biol. 2019 Apr;188:86-89. doi: 10.1016/j.jsbmb.2018.12.009. Epub 2018 Dec 23. J Steroid Biochem Mol Biol. 2019. PMID: 30586624 Review.
Cited by
-
Serum neurofilament light chain is increased in hereditary spastic paraplegias.Ann Clin Transl Neurol. 2018 May 21;5(7):876-882. doi: 10.1002/acn3.583. eCollection 2018 Jul. Ann Clin Transl Neurol. 2018. PMID: 30009206 Free PMC article.
-
Exome Sequencing Reveals a Novel Homozygous Frameshift Mutation in the CYP7B1 Gene in a Japanese Patient with SPG5.Intern Med. 2019 Mar 1;58(5):719-722. doi: 10.2169/internalmedicine.1839-18. Epub 2018 Oct 17. Intern Med. 2019. PMID: 30333426 Free PMC article.
-
Chenodeoxycholic acid rescues axonal degeneration in induced pluripotent stem cell-derived neurons from spastic paraplegia type 5 and cerebrotendinous xanthomatosis patients.Orphanet J Rare Dis. 2023 Apr 6;18(1):72. doi: 10.1186/s13023-023-02666-w. Orphanet J Rare Dis. 2023. PMID: 37024986 Free PMC article.
-
High levels of 27-hydroxycholesterol results in synaptic plasticity alterations in the hippocampus.Sci Rep. 2021 Feb 12;11(1):3736. doi: 10.1038/s41598-021-83008-3. Sci Rep. 2021. PMID: 33580102 Free PMC article.
-
Oxysterols as lipid mediators: Their biosynthetic genes, enzymes and metabolites.Prostaglandins Other Lipid Mediat. 2020 Apr;147:106381. doi: 10.1016/j.prostaglandins.2019.106381. Epub 2019 Nov 4. Prostaglandins Other Lipid Mediat. 2020. PMID: 31698146 Free PMC article. Review.
References
-
- Arnoldi A, Crimella C, Tenderini E, Martinuzzi A, D'Angelo MG, Musumeci O, et al.Clinical phenotype variability in patients with hereditary spastic paraplegia type 5 associated with CYP7B1 mutations. Clin Genet 2012; 81: 150–7. - PubMed
-
- Babiker A, Dzeletovic S, Wiklund B, Pettersson N, Salonen J, Nyyssonen K, et al.Patients with atherosclerosis may have increased circulating levels of 27-hydroxycholesterol and cholestenoic acid. Scand J Clin Lab Invest 2005; 65: 365–75. - PubMed
-
- Biancheri R, Ciccolella M, Rossi A, Tessa A, Cassandrini D, Minetti C, et al.White matter lesions in spastic paraplegia with mutations in SPG5/CYP7B1. Neuromuscul Disord 2009; 19: 62–5. - PubMed
-
- Bjorkhem I, Falk O. Assay of the major bile acids in serum by isotope dilution-mass spectrometry. Scand J Clin Lab Invest 1983; 43: 163–70. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
