

Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B
- PMID: 29127144
- PMCID: PMC5726601
- DOI: 10.4049/jimmunol.1700460
Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B
Abstract
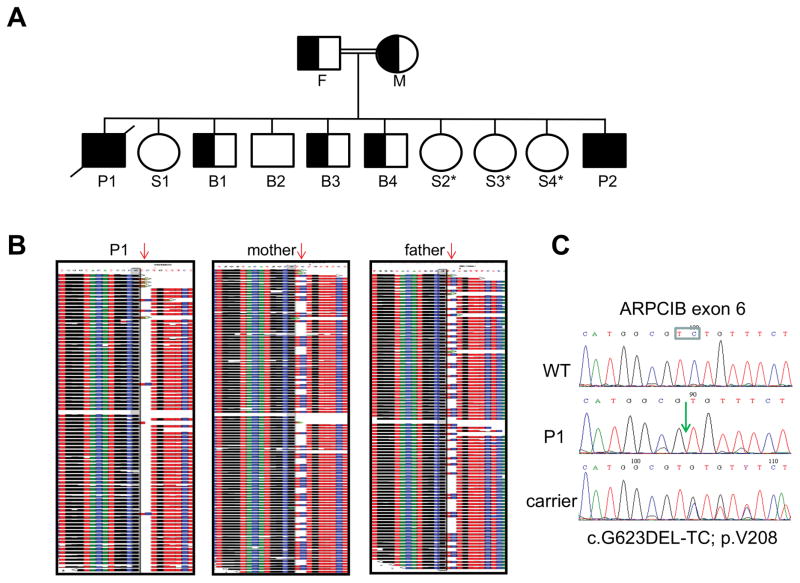
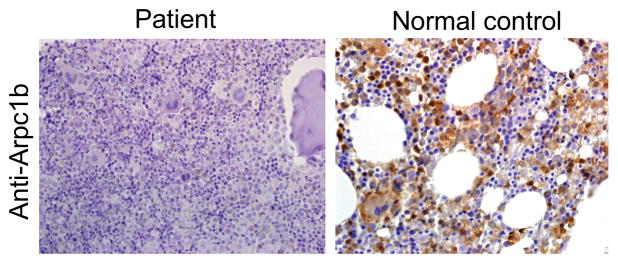
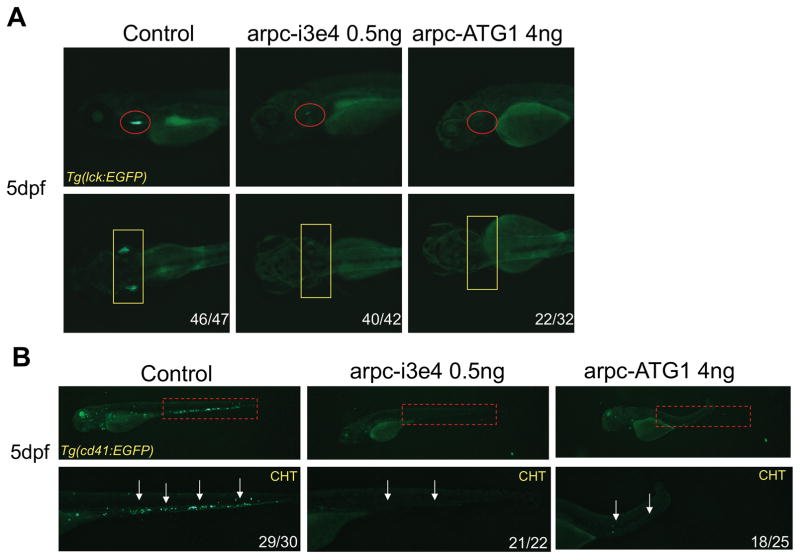
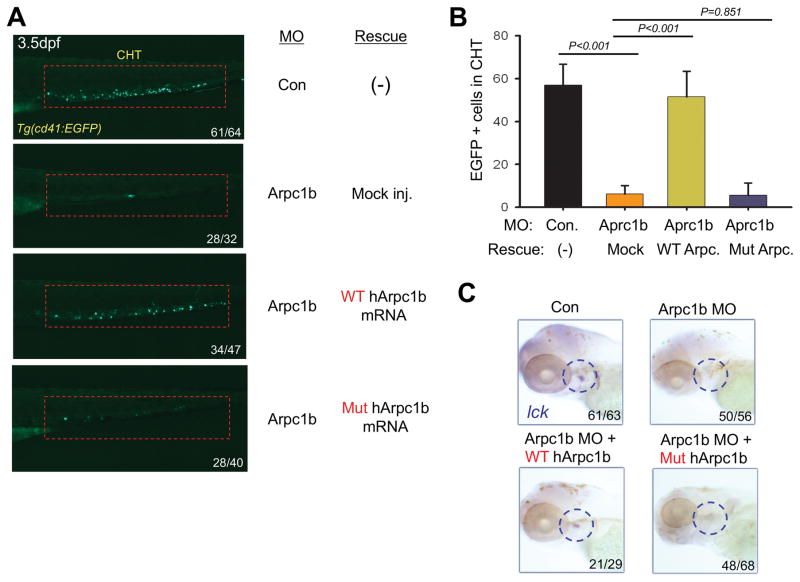
Regulation of the actin cytoskeleton is crucial for normal development and function of the immune system, as evidenced by the severe immune abnormalities exhibited by patients bearing inactivating mutations in the Wiskott-Aldrich syndrome protein (WASP), a key regulator of actin dynamics. WASP exerts its effects on actin dynamics through a multisubunit complex termed Arp2/3. Despite the critical role played by Arp2/3 as an effector of WASP-mediated control over actin polymerization, mutations in protein components of the Arp2/3 complex had not previously been identified as a cause of immunodeficiency. Here, we describe two brothers with hematopoietic and immunologic symptoms reminiscent of Wiskott-Aldrich syndrome (WAS). However, these patients lacked mutations in any of the genes previously associated with WAS. Whole-exome sequencing revealed a homozygous 2 bp deletion, n.c.G623DEL-TC (p.V208VfsX20), in Arp2/3 complex component ARPC1B that causes a frame shift resulting in premature termination. Modeling of the disease in zebrafish revealed that ARPC1B plays a critical role in supporting T cell and thrombocyte development. Moreover, the defects in development caused by ARPC1B loss could be rescued by the intact human ARPC1B ortholog, but not by the p.V208VfsX20 variant identified in the patients. Moreover, we found that the expression of ARPC1B is restricted to hematopoietic cells, potentially explaining why a mutation in ARPC1B has now been observed as a cause of WAS, whereas mutations in other, more widely expressed, components of the Arp2/3 complex have not been observed.
Copyright © 2017 by The American Association of Immunologists, Inc.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Derry JM, Ochs HD, Francke U. Isolation of a novel gene mutated in Wiskott-Aldrich syndrome. Cell. 1994;78:635–644. - PubMed
-
- Chandra S, Bronicki L, Nagaraj CB, Zhang K. WAS-Related Disorders. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Ledbetter N, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R) Seattle (WA): 1993. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
