

Review
doi: 10.1007/978-1-4939-7299-9_3.
Current Standards of Care and Long Term Outcomes for Thalassemia and Sickle Cell Disease
Affiliations
- PMID: 29127677
- PMCID: PMC5720159
- DOI: 10.1007/978-1-4939-7299-9_3
Item in Clipboard
Review
Current Standards of Care and Long Term Outcomes for Thalassemia and Sickle Cell Disease
Adv Exp Med Biol.
2017.
Abstract
Thalassemia and sickle cell disease (SCD) are disorders of hemoglobin that affect millions of people worldwide. The carrier states for these diseases arose as common, balanced polymorphisms during human history because they afforded protection against severe forms of malaria. These complex, multisystem diseases are reviewed here with a focus on current standards of clinical management and recent research findings. The importance of a comprehensive, multidisciplinary and lifelong system of care is also emphasized.
Keywords: Clinical care; Outcome; Sickle cell disease; Standard care; Thalassaemia; Thalassemia.
Figures
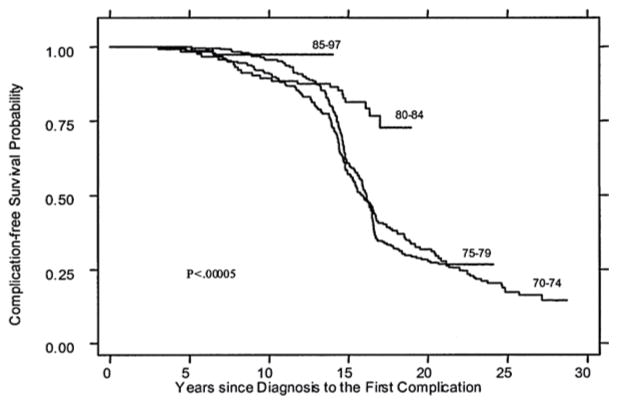
Improving survival in thalassemia major. Complication-free survival for patients with thalassemia major cared for in Italy is shown by birth cohort (1970–74, 1975–79, 1980–84, 1985–97). From: Borgna-Pignatti C, et al. Ann N Y Acad Sci. 2005;1054(1):40–7. Reprinted with permission from John Wiley and Sons
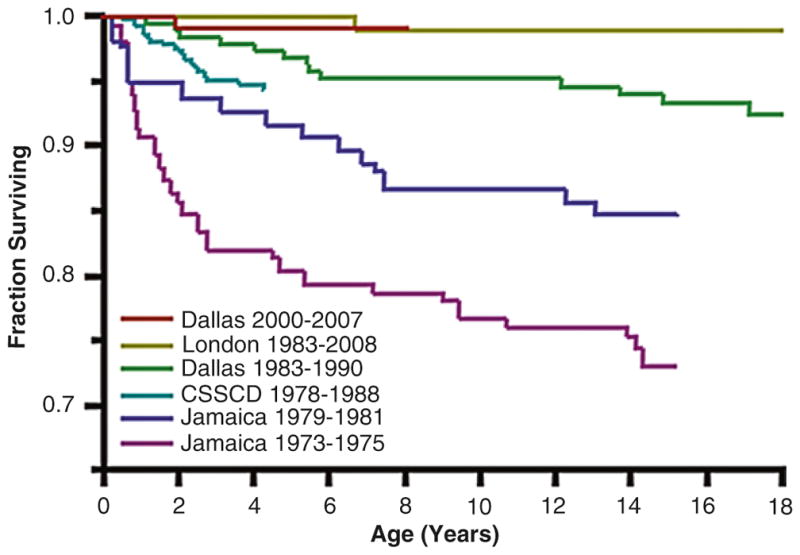
Improving survival in sickle cell disease (SCD). Overall survival curves for patients with sickle cell anemia (Hb SS) and sickle-β0-thalassemia spanning by years of birth are shown for large SCD cohorts in the US, UK and Jamaica. Figure adapted from: Quinn et al. Blood. 2010;115:3447–3452
References
-
- Chui DH, Hardison R, Riemer C, et al. An electronic database of human hemoglobin variants on the World Wide Web. Blood. 1998;91(8):2643–2644. - PubMed
-
- Laig M, Pape M, Hundrieser J, et al. The distribution of the Hb constant spring gene in Southeast Asian populations. Hum Genet. 1990;84(2):188–190. - PubMed
-
- Voon HPJ, Vadolas J. Controlling alpha-globin: a review of alpha-globin expression and its impact on beta-thalassemia. Haematologica. 2008;93(12):1868–1876. - PubMed
-
- Pootrakul P, Sirankapracha P, Hemsorach S, et al. A correlation of erythrokinetics, ineffective erythropoiesis, and erythroid precursor apoptosis in thai patients with thalassemia. Blood. 2000;96(7):2606–2612. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
