

Readers of DNA methylation, the MBD family as potential therapeutic targets
- PMID: 29128342
- PMCID: PMC6438182
- DOI: 10.1016/j.pharmthera.2017.11.002
Readers of DNA methylation, the MBD family as potential therapeutic targets
Erratum in
-
Corrigendum to "Readers of DNA methylation, the MBD family as potential therapeutic targets" [Pharmacology & Therapeutics 184 (1) (2018) 98-111].Pharmacol Ther. 2018 Oct;190:237-238. doi: 10.1016/j.pharmthera.2018.09.006. Pharmacol Ther. 2018. PMID: 30268235 No abstract available.
Abstract
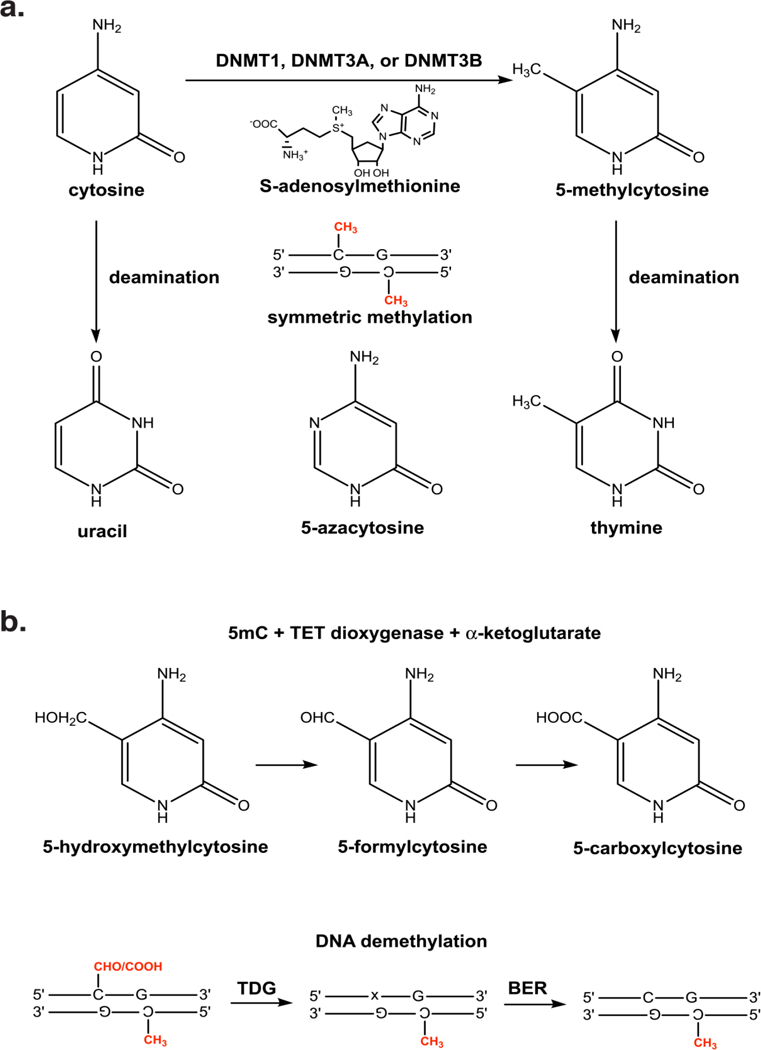
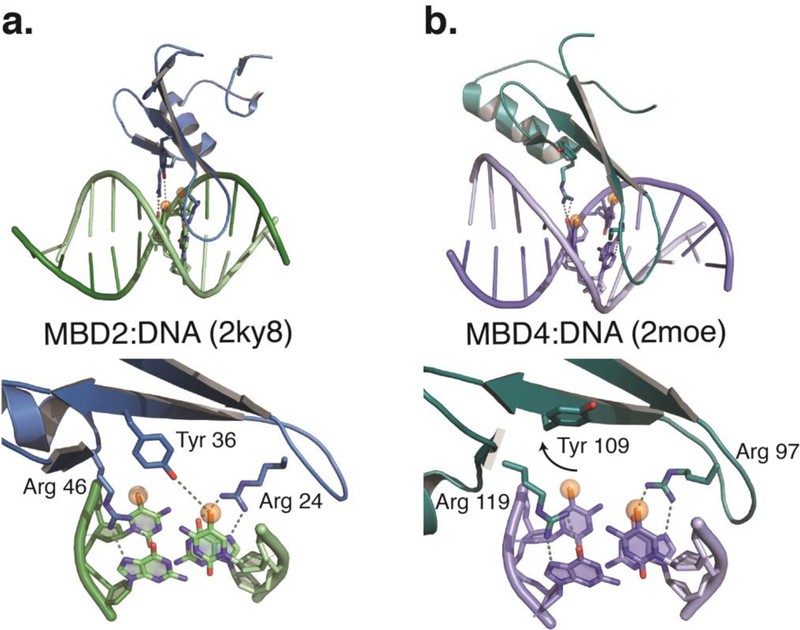
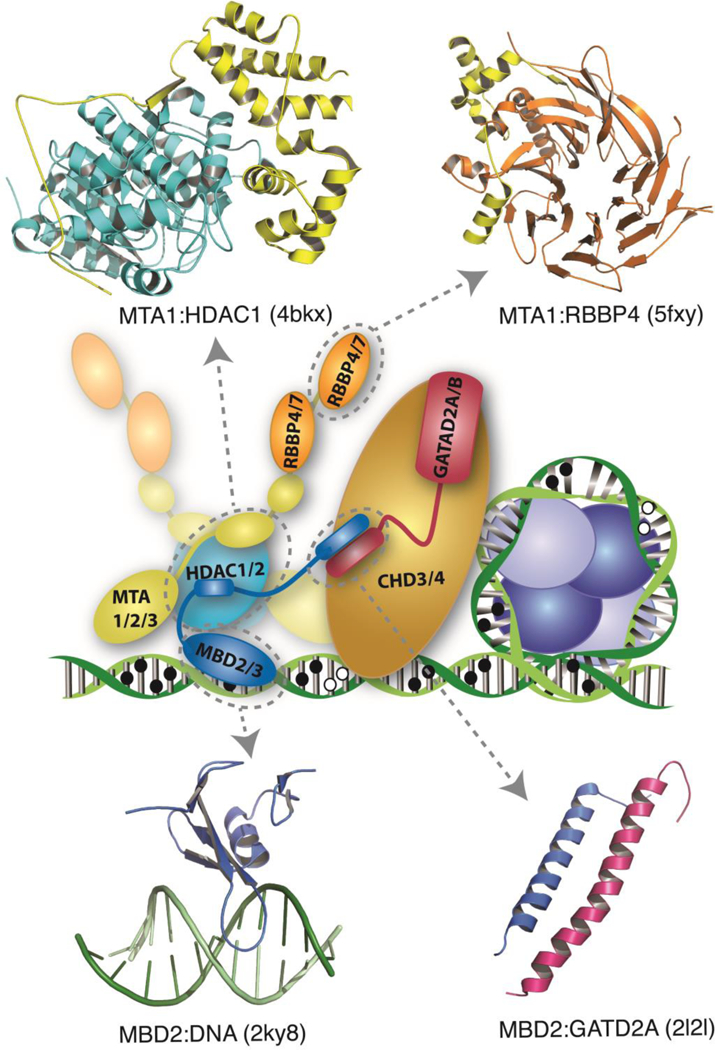
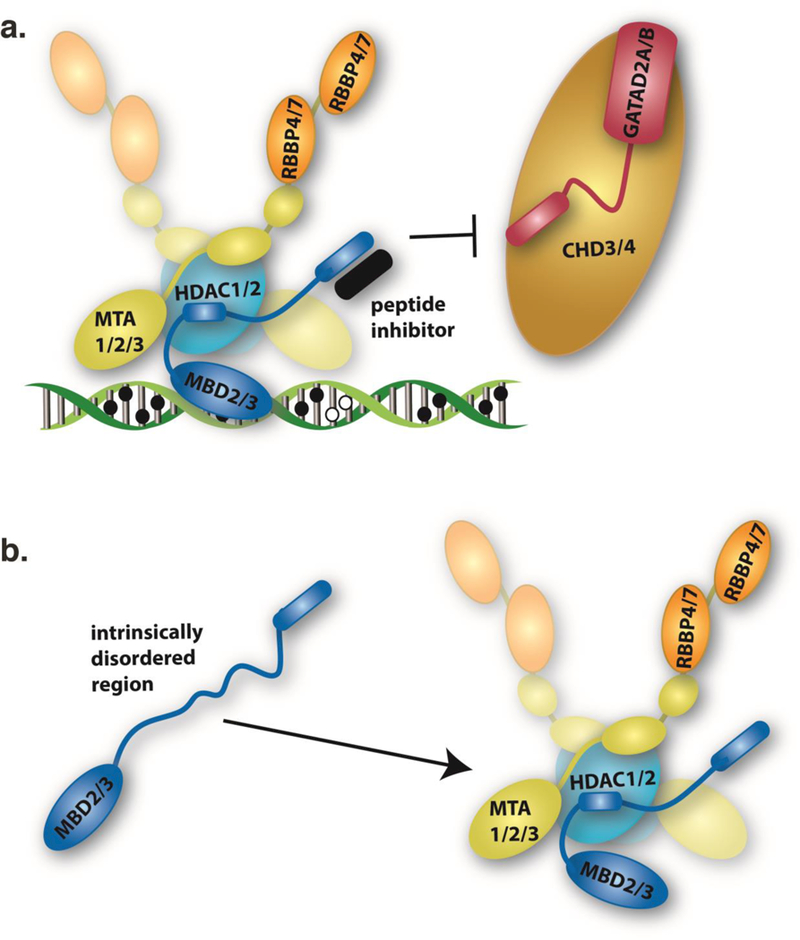
DNA methylation represents a fundamental epigenetic modification that regulates chromatin architecture and gene transcription. Many diseases, including cancer, show aberrant methylation patterns that contribute to the disease phenotype. DNA methylation inhibitors have been used to block methylation dependent gene silencing to treat hematopoietic neoplasms and to restore expression of developmentally silenced genes. However, these inhibitors disrupt methylation globally and show significant off-target toxicities. As an alternative approach, we have been studying readers of DNA methylation, the 5-methylcytosine binding domain family of proteins, as potential therapeutic targets to restore expression of aberrantly and developmentally methylated and silenced genes. In this review, we discuss the role of DNA methylation in gene regulation and cancer development, the structure and function of the 5-methylcytosine binding domain family of proteins, and the possibility of targeting the complexes these proteins form to treat human disease.
Keywords: 5-Methylcytosine binding domain; Chromatin; DNA methylation; Gene regulation; NuRD.
Copyright © 2017 Elsevier Inc. All rights reserved.
Conflict of interest statement
Figures
References
-
- Aguilera C, Nakagawa K, Sancho R, Chakraborty A, Hendrich B, & Behrens A (2011). c-Jun N-terminal phosphorylation antagonises recruitment of the Mbd3/NuRD repressor complex. Nature, 469, 231–235. - PubMed
-
- Alqarni SS, Murthy A, Zhang W, Przewloka MR, Silva AP, Watson AA, Lejon S, Pei XY, Smits AH, Kloet SL, Wang H, Shepherd NE, Stokes PH, Blobel GA, Vermeulen M, Glover DM, Mackay JP, & Laue ED (2014). Insight into the architecture of the NuRD complex: structure of the RbAp48-MTA1 subcomplex. J Biol Chem, 289, 21844–21855. - PMC - PubMed
-
- Amir RE, Van den Veyver IB, Wan M, Tran CQ, Francke U, & Zoghbi HY (1999). Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat Genet, 23, 185–188. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
