

Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones
- PMID: 29130686
- PMCID: PMC7744120
- DOI: 10.1021/jacs.7b08417
Thioamide Substitution Selectively Modulates Proteolysis and Receptor Activity of Therapeutic Peptide Hormones
Abstract
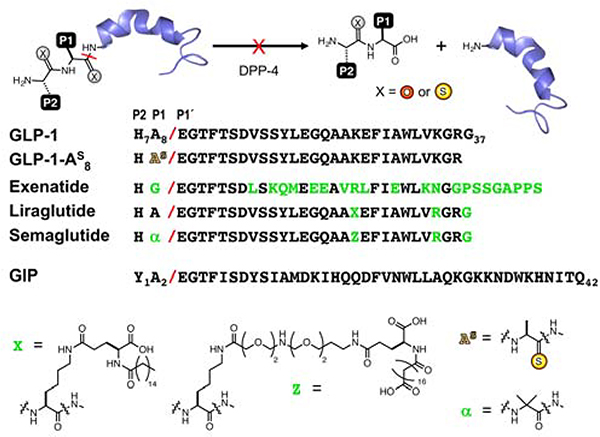
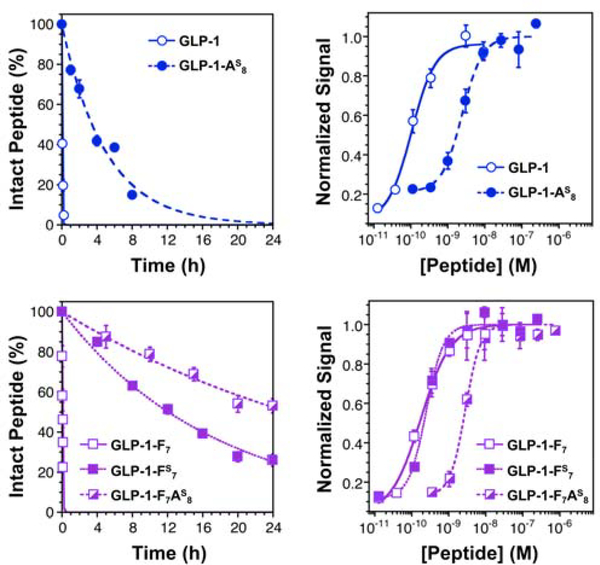
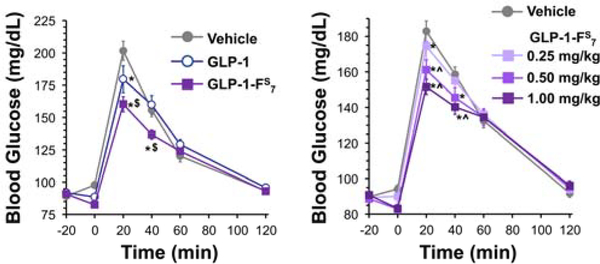
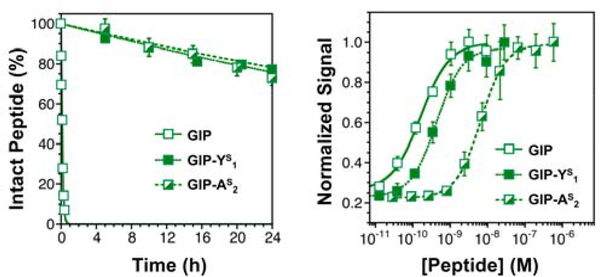
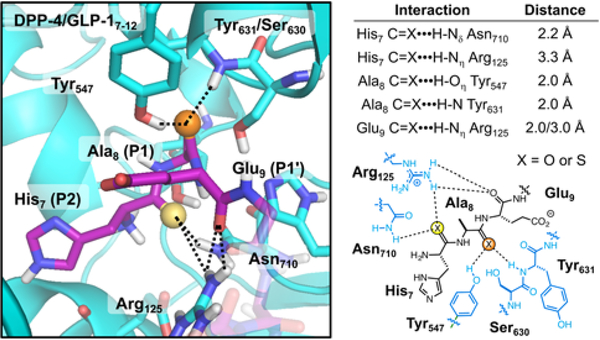
Peptide hormones are attractive as injectable therapeutics and imaging agents, but they often require extensive modification by mutagenesis and/or chemical synthesis to prevent rapid in vivo degradation. Alternatively, the single-atom, O-to-S modification of peptide backbone thioamidation has the potential to selectively perturb interactions with proteases while preserving interactions with other proteins, such as target receptors. Here, we use the validated diabetes therapeutic, glucagon-like peptide-1 (GLP-1), and the target of clinical investigation, gastric inhibitory polypeptide (GIP), as proof-of-principle peptides to demonstrate the value of thioamide substitution. In GLP-1 and GIP, a single thioamide near the scissile bond renders these peptides up to 750-fold more stable than the corresponding oxopeptides toward cleavage by dipeptidyl peptidase 4, the principal regulator of their in vivo stability. These stabilized analogues are nearly equipotent with their parent peptide in cyclic AMP activation assays, but the GLP-1 thiopeptides have much lower β-arrestin potency, making them novel agonists with altered signaling bias. Initial tests show that a thioamide GLP-1 analogue is biologically active in rats, with an in vivo potency for glycemic control surpassing that of native GLP-1. Taken together, these experiments demonstrate the potential for thioamides to modulate specific protein interactions to increase proteolytic stability or tune activation of different signaling pathways.
Figures
References
-
- Mullard A, Nat. Rev. Drug Discov. 2013, 12 (5), 329–332. - PubMed
-
- Projan SJ; Gill D; Lu Z; Herrmann SH, Expert Opin. Biol. Ther. 2004, 4 (8), 1345–1350. - PubMed
-
- Verdine GL; Walensky LD, Clin. Cancer Res. 2007, 13 (24), 7264–7270. - PubMed
-
- Buse JB; Rosenstock J; Sesti G; Schmidt WE; Montanya E; Brett JH; Zychma M; Blonde L, Lancet 2009, 374 (9683), 39–47. - PubMed
-
- Kreymann B; Ghatei M; Williams G; Bloom S, Lancet 1987, 330 (8571), 1300–1304. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
