

Leukocyte RhoA exchange factor Arhgef1 mediates vascular inflammation and atherosclerosis
- PMID: 29130930
- PMCID: PMC5707138
- DOI: 10.1172/JCI92702
Leukocyte RhoA exchange factor Arhgef1 mediates vascular inflammation and atherosclerosis
Abstract
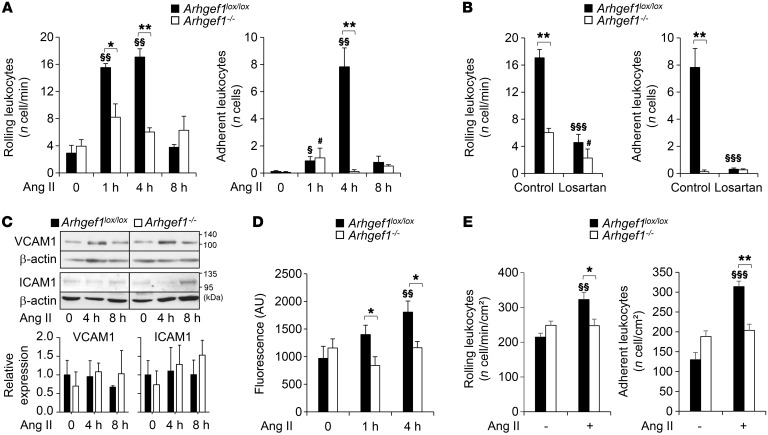
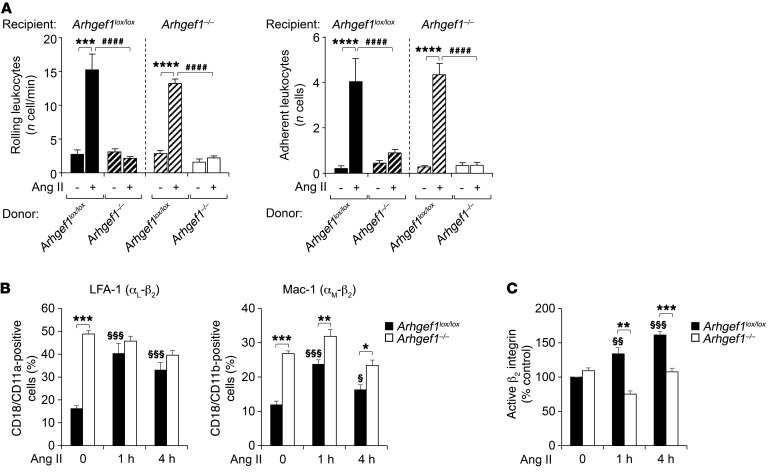
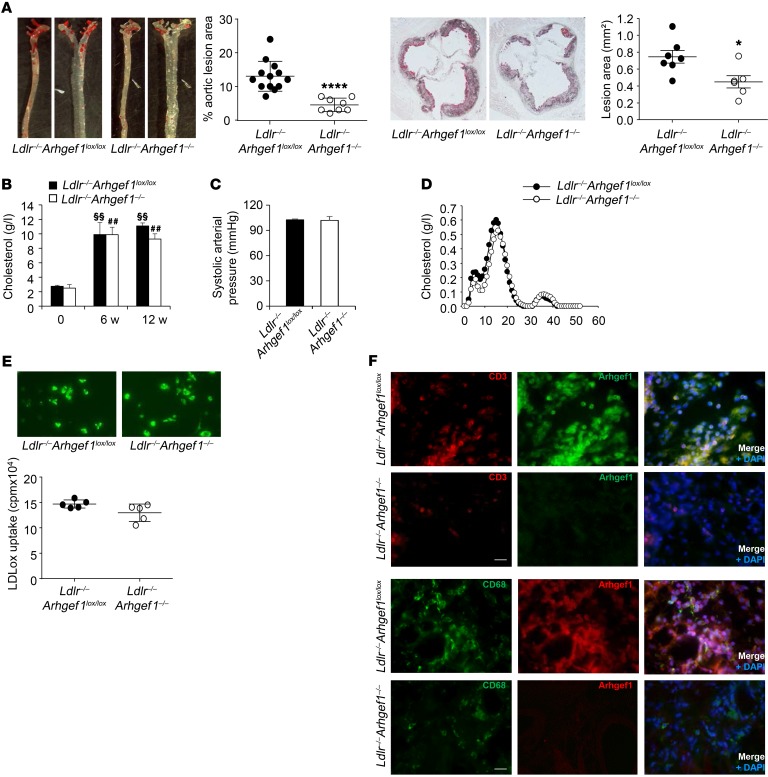
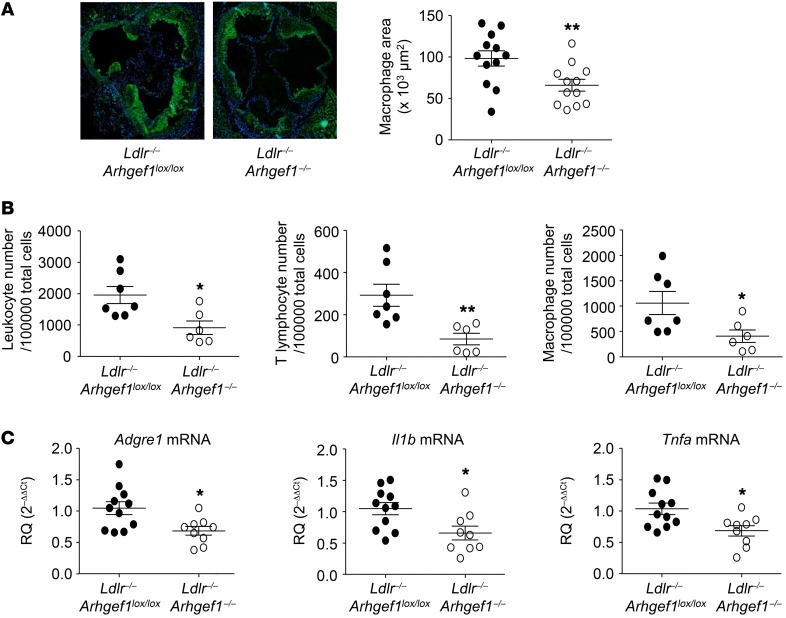
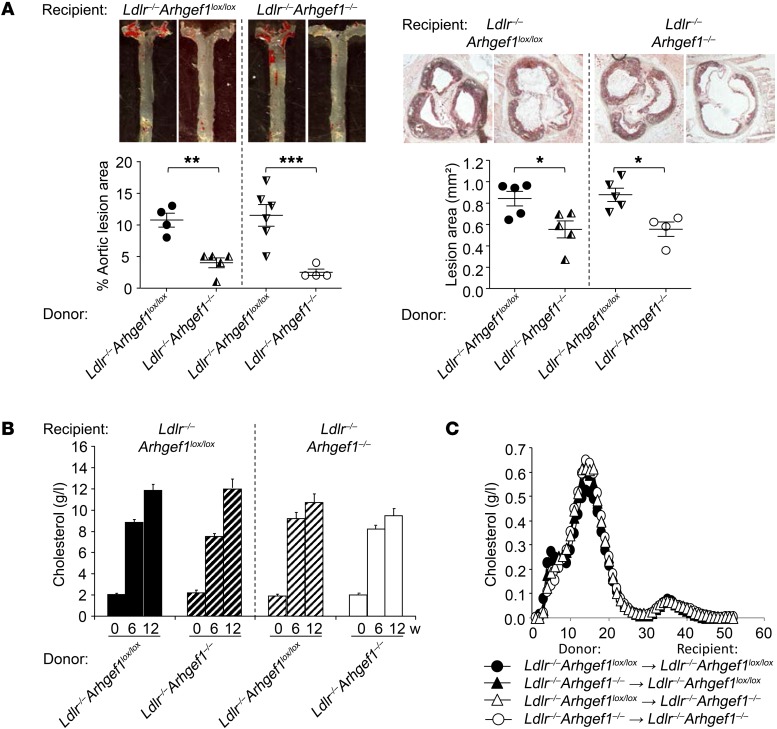
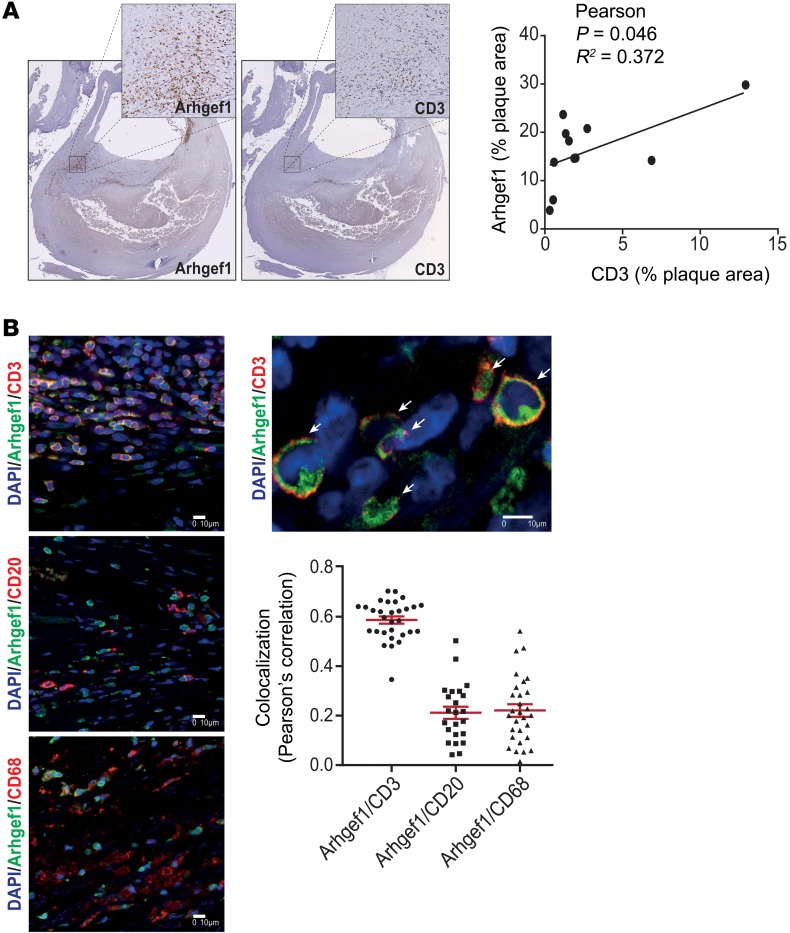
Abnormal activity of the renin-angiotensin-aldosterone system plays a causal role in the development of hypertension, atherosclerosis, and associated cardiovascular events such as myocardial infarction, stroke, and heart failure. As both a vasoconstrictor and a proinflammatory mediator, angiotensin II (Ang II) is considered a potential link between hypertension and atherosclerosis. However, a role for Ang II-induced inflammation in atherosclerosis has not been clearly established, and the molecular mechanisms and intracellular signaling pathways involved are not known. Here, we demonstrated that the RhoA GEF Arhgef1 is essential for Ang II-induced inflammation. Specifically, we showed that deletion of Arhgef1 in a murine model prevents Ang II-induced integrin activation in leukocytes, thereby preventing Ang II-induced recruitment of leukocytes to the endothelium. Mice lacking both LDL receptor (LDLR) and Arhgef1 were protected from high-fat diet-induced atherosclerosis. Moreover, reconstitution of Ldlr-/- mice with Arhgef1-deficient BM prevented high-fat diet-induced atherosclerosis, while reconstitution of Ldlr-/- Arhgef1-/- with WT BM exacerbated atherosclerotic lesion formation, supporting Arhgef1 activation in leukocytes as causal in the development of atherosclerosis. Thus, our data highlight the importance of Arhgef1 in cardiovascular disease and suggest targeting Arhgef1 as a potential therapeutic strategy against atherosclerosis.
Keywords: Atherosclerosis; G-proteins; Integrins; Vascular Biology.
Conflict of interest statement
Figures
References
-
- de Gasparo M, Catt KJ, Inagami T, Wright JW, Unger T. International union of pharmacology. XXIII. The angiotensin II receptors. Pharmacol Rev. 2000;52(3):415–472. - PubMed
-
- Cassis LA, Rateri DL, Lu H, Daugherty A. Bone marrow transplantation reveals that recipient AT1a receptors are required to initiate angiotensin II-induced atherosclerosis and aneurysms. Arterioscler Thromb Vasc Biol. 2007;27(2):380–386. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
