

Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia
- PMID: 29131699
- PMCID: PMC5756322
- DOI: 10.1200/JCO.2017.74.0449
Genotype-Specific Minimal Residual Disease Interpretation Improves Stratification in Pediatric Acute Lymphoblastic Leukemia
Abstract
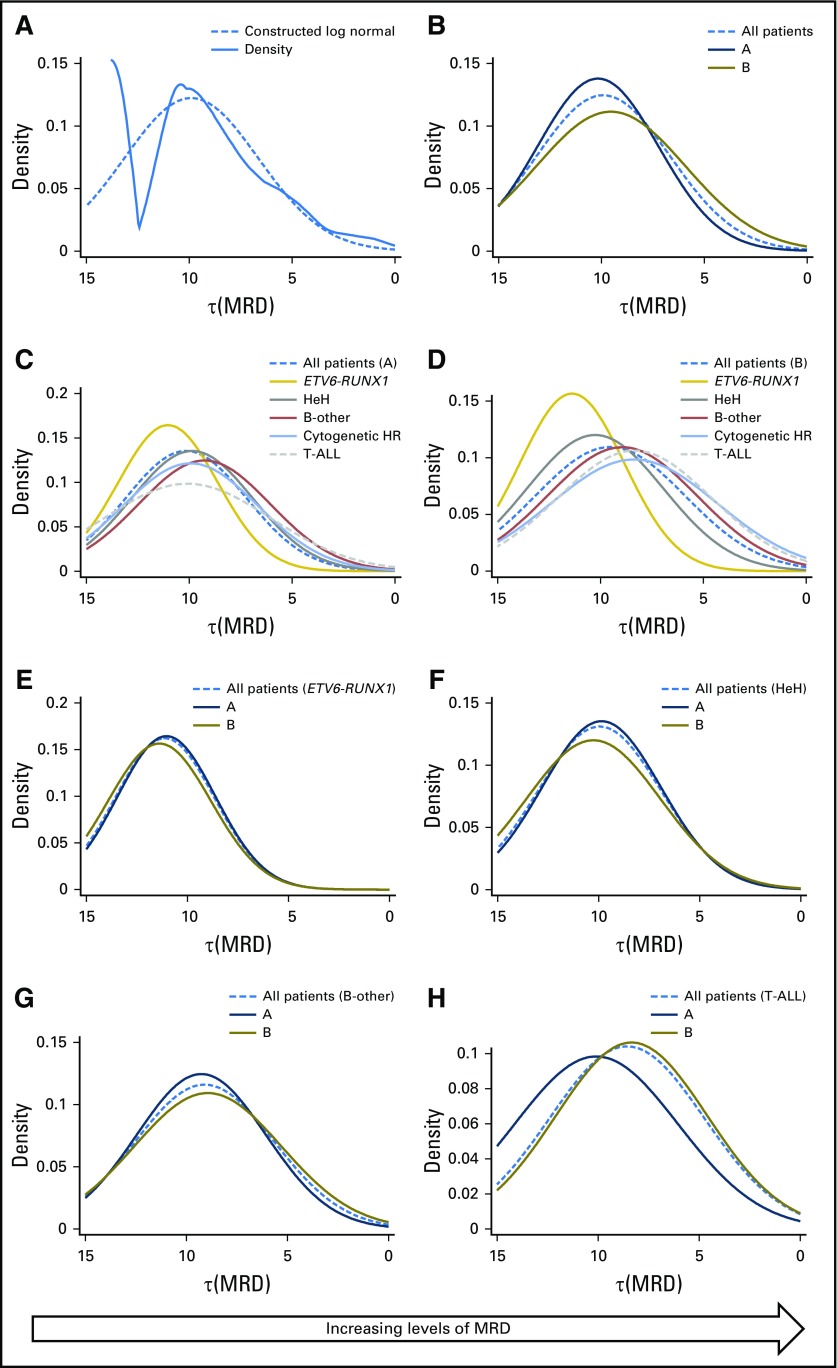
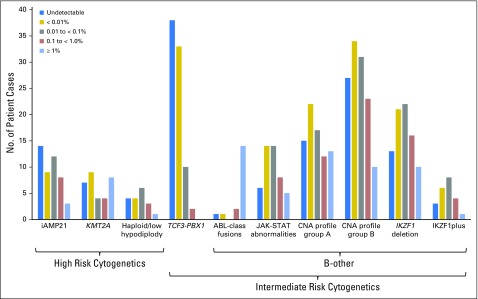
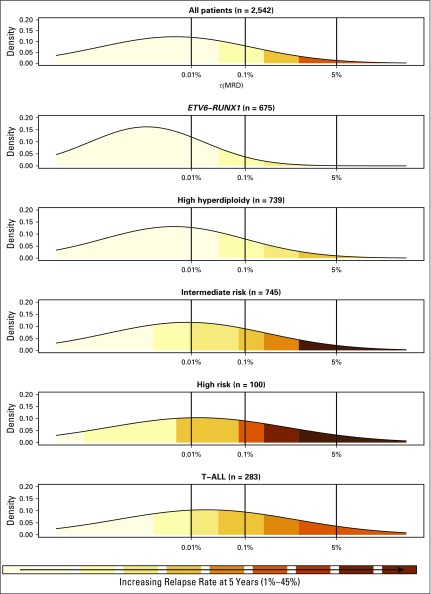
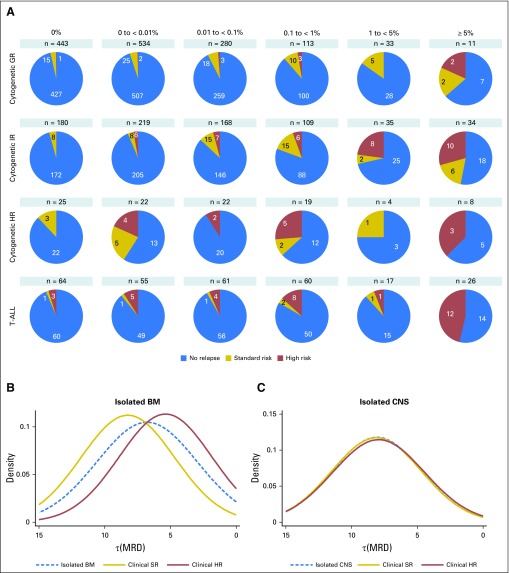
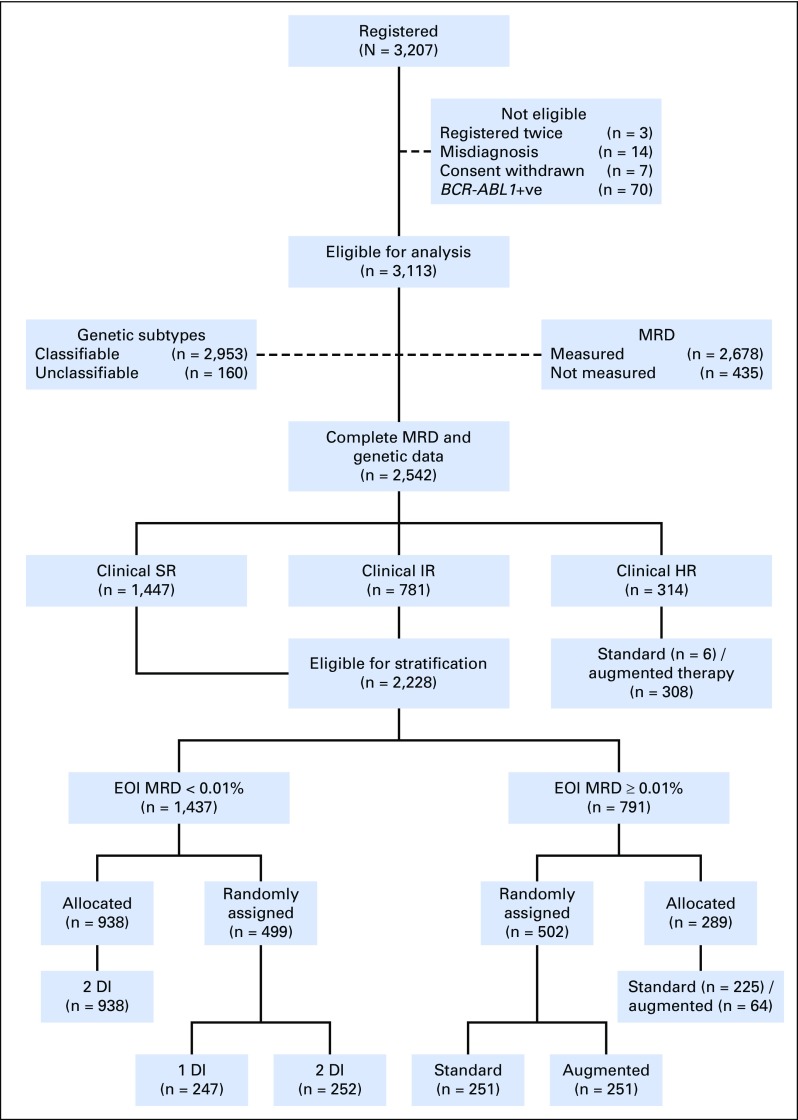
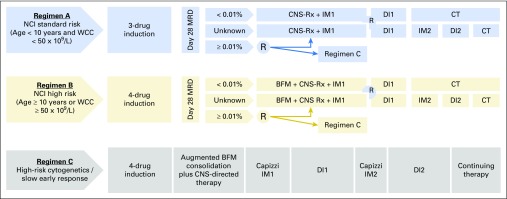
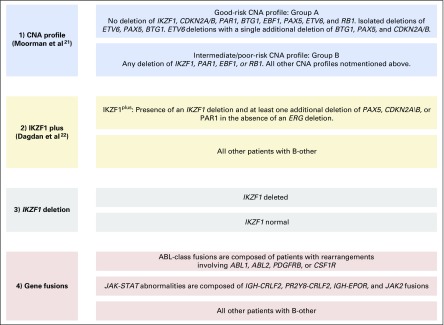
Purpose Minimal residual disease (MRD) and genetic abnormalities are important risk factors for outcome in acute lymphoblastic leukemia. Current risk algorithms dichotomize MRD data and do not assimilate genetics when assigning MRD risk, which reduces predictive accuracy. The aim of our study was to exploit the full power of MRD by examining it as a continuous variable and to integrate it with genetics. Patients and Methods We used a population-based cohort of 3,113 patients who were treated in UKALL2003, with a median follow-up of 7 years. MRD was evaluated by polymerase chain reaction analysis of Ig/TCR gene rearrangements, and patients were assigned to a genetic subtype on the basis of immunophenotype, cytogenetics, and fluorescence in situ hybridization. To examine response kinetics at the end of induction, we log-transformed the absolute MRD value and examined its distribution across subgroups. Results MRD was log normally distributed at the end of induction. MRD distributions of patients with distinct genetic subtypes were different ( P < .001). Patients with good-risk cytogenetics demonstrated the fastest disease clearance, whereas patients with high-risk genetics and T-cell acute lymphoblastic leukemia responded more slowly. The risk of relapse was correlated with MRD kinetics, and each log reduction in disease level reduced the risk by 20% (hazard ratio, 0.80; 95% CI, 0.77 to 0.83; P < .001). Although the risk of relapse was directly proportional to the MRD level within each genetic risk group, absolute relapse rate that was associated with a specific MRD value or category varied significantly by genetic subtype. Integration of genetic subtype-specific MRD values allowed more refined risk group stratification. Conclusion A single threshold for assigning patients to an MRD risk group does not reflect the response kinetics of the different genetic subtypes. Future risk algorithms should integrate genetics with MRD to accurately identify patients with the lowest and highest risk of relapse.
Figures
Comment in
-
Integrated Risk Stratification Using Minimal Residual Disease and Sentinel Genetic Alterations in Pediatric Acute Lymphoblastic Leukemia.J Clin Oncol. 2018 Jan 1;36(1):4-6. doi: 10.1200/JCO.2017.76.0504. Epub 2017 Nov 13. J Clin Oncol. 2018. PMID: 29131700 No abstract available.
References
-
- Vora A, Goulden N, Wade R, et al. : Treatment reduction for children and young adults with low-risk acute lymphoblastic leukaemia defined by minimal residual disease (UKALL 2003): A randomised controlled trial. Lancet Oncol 14:199-209, 2013 - PubMed
-
- Coustan-Smith E, Sancho J, Hancock ML, et al. : Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood 96:2691-2696, 2000 - PubMed
-
- Bartram J, Wade R, Vora A, et al. : Excellent outcome of minimal residual disease-defined low-risk patients is sustained with more than 10 years follow-up: Results of UK paediatric acute lymphoblastic leukaemia trials 1997-2003. Arch Dis Child 101:449-454, 2016 - PubMed
-
- Vora A, Goulden N, Mitchell C, et al. : Augmented post-remission therapy for a minimal residual disease-defined high-risk subgroup of children and young people with clinical standard-risk and intermediate-risk acute lymphoblastic leukaemia (UKALL 2003): A randomised controlled trial. Lancet Oncol 15:809-818, 2014 - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
