

Excessive aggregation of membrane proteins in the Martini model
- PMID: 29131844
- PMCID: PMC5683612
- DOI: 10.1371/journal.pone.0187936
Excessive aggregation of membrane proteins in the Martini model
Abstract
The coarse-grained Martini model is employed extensively to study membrane protein oligomerization. While this approach is exceptionally promising given its computational efficiency, it is alarming that a significant fraction of these studies demonstrate unrealistic protein clusters, whose formation is essentially an irreversible process. This suggests that the protein-protein interactions are exaggerated in the Martini model. If this held true, then it would limit the applicability of Martini to study multi-protein complexes, as the rapidly clustering proteins would not be able to properly sample the correct dimerization conformations. In this work we first demonstrate the excessive protein aggregation by comparing the dimerization free energies of helical transmembrane peptides obtained with the Martini model to those determined from FRET experiments. Second, we show that the predictions provided by the Martini model for the structures of transmembrane domain dimers are in poor agreement with the corresponding structures resolved using NMR. Next, we demonstrate that the first issue can be overcome by slightly scaling down the Martini protein-protein interactions in a manner, which does not interfere with the other Martini interaction parameters. By preventing excessive, irreversible, and non-selective aggregation of membrane proteins, this approach renders the consideration of lateral dynamics and protein-lipid interactions in crowded membranes by the Martini model more realistic. However, this adjusted model does not lead to an improvement in the predicted dimer structures. This implicates that the poor agreement between the Martini model and NMR structures cannot be cured by simply uniformly reducing the interactions between all protein beads. Instead, a careful amino-acid specific adjustment of the protein-protein interactions is likely required.
Conflict of interest statement
Figures
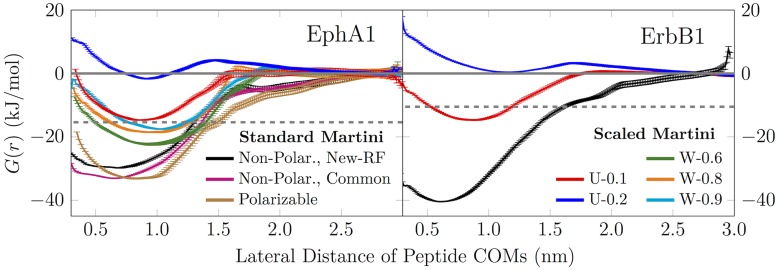
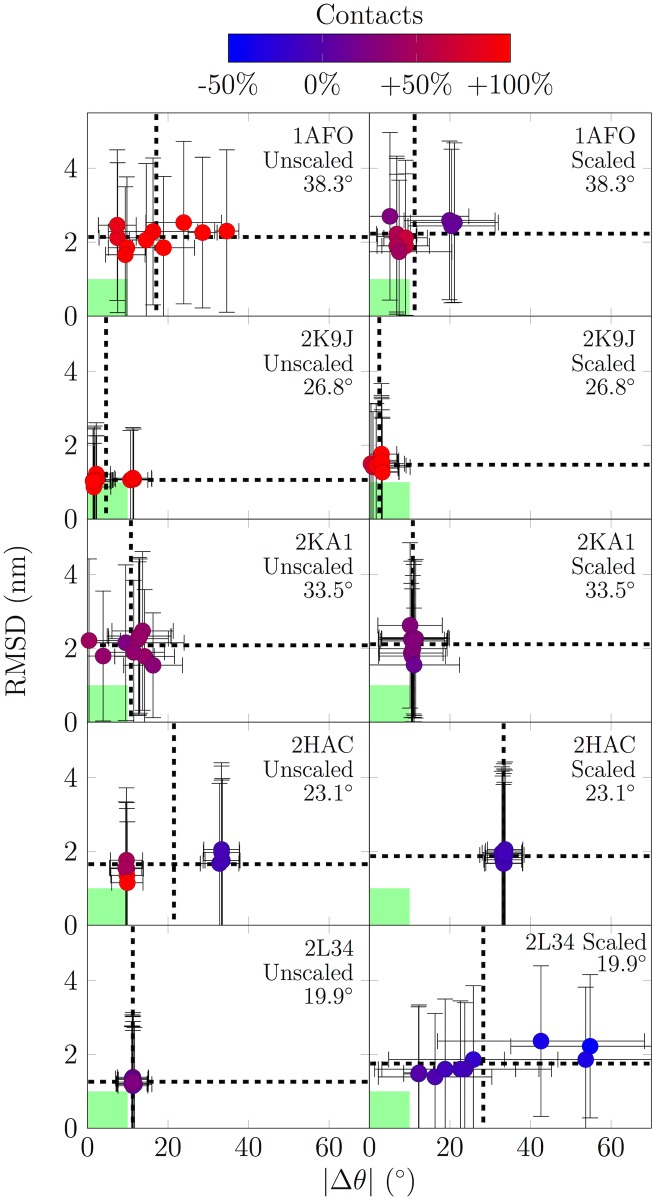
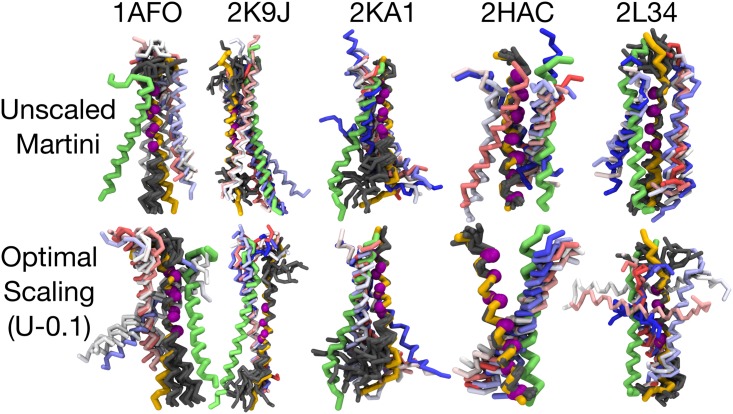
References
-
- Ferré S, Casadó V, Devi LA, Filizola M, Jockers R, Lohse MJ, et al. G Protein–Coupled Receptor Oligomerization Revisited: Functional and Pharmacological Perspectives. Pharmacol Rev. 2014;66(2):413–434. doi: 10.1124/pr.113.008052 - DOI - PMC - PubMed
-
- Gomes I, Ayoub MA, Fujita W, Jaeger WC, Pfleger KD, Devi LA. G Protein-Coupled Receptor Heteromers. Annu Rev Pharmacol Toxicol. 2016;56:403–425. doi: 10.1146/annurev-pharmtox-011613-135952 - DOI - PMC - PubMed
-
- Haass C, Selkoe DJ. Soluble Protein Oligomers in Neurodegeneration: Lessons From the Alzheimer’s Amyloid β-Peptide. Nat Rev Mol Cell Biol. 2007;8(2):101–112. doi: 10.1038/nrm2101 - DOI - PubMed
-
- Ghosh A, Sonavane U, Joshi R. Multiscale Modelling to Understand the Self-Assembly Mechanism of Human β2-Adrenergic Receptor in Lipid Bilayer. Computat Biol Chem. 2014;48:29–39. doi: 10.1016/j.compbiolchem.2013.11.002 - DOI - PubMed
-
- Mondal S, Johnston JM, Wang H, Khelashvili G, Filizola M, Weinstein H. Membrane Driven Spatial Organization of GPCRs. Sci Rep. 2013;3:2909 doi: 10.1038/srep02909 - DOI - PMC - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
