

A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy
- PMID: 29132144
- PMCID: PMC6137806
- DOI: 10.1038/nature24473
A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy
Abstract
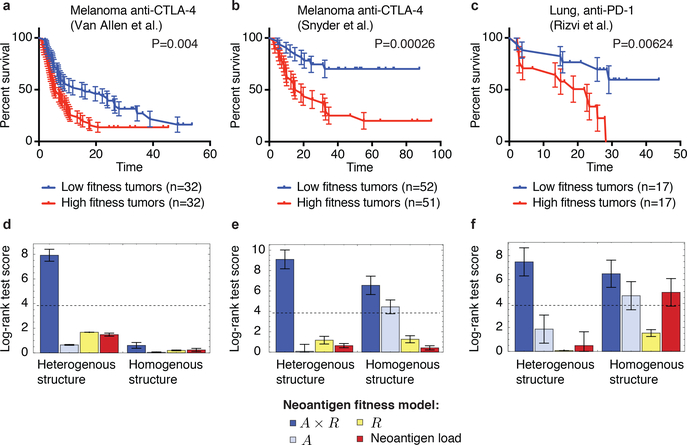
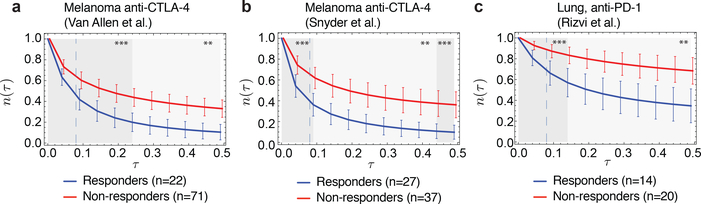
Checkpoint blockade immunotherapies enable the host immune system to recognize and destroy tumour cells. Their clinical activity has been correlated with activated T-cell recognition of neoantigens, which are tumour-specific, mutated peptides presented on the surface of cancer cells. Here we present a fitness model for tumours based on immune interactions of neoantigens that predicts response to immunotherapy. Two main factors determine neoantigen fitness: the likelihood of neoantigen presentation by the major histocompatibility complex (MHC) and subsequent recognition by T cells. We estimate these components using the relative MHC binding affinity of each neoantigen to its wild type and a nonlinear dependence on sequence similarity of neoantigens to known antigens. To describe the evolution of a heterogeneous tumour, we evaluate its fitness as a weighted effect of dominant neoantigens in the subclones of the tumour. Our model predicts survival in anti-CTLA-4-treated patients with melanoma and anti-PD-1-treated patients with lung cancer. Importantly, low-fitness neoantigens identified by our method may be leveraged for developing novel immunotherapies. By using an immune fitness model to study immunotherapy, we reveal broad similarities between the evolution of tumours and rapidly evolving pathogens.
Conflict of interest statement
Conflicts of interest
M.Ł. has consulted for Merck. V.P.B. has received research funding from Bristol- Myers Squibb. A.J.L. is on the board of directors for Adaptive Biotechnologies and has consulted for Jansen pharmaceuticals and Merck. T.A.C. is a co-founder of Gritstone Oncology and holds equity. T.A.C. receives grant funding from Bristol Myers Squibb. N.A.R is co-founder and shareholder of Gritstone Oncology. M.D.H has consulted for Genentech, BMS, Merck, AstraZeneca, Janssen, Novartis. B.D.G. has consulted for Merck.
Figures
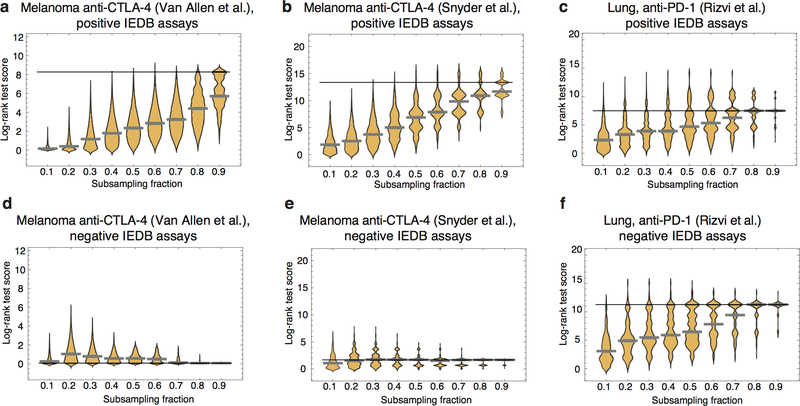
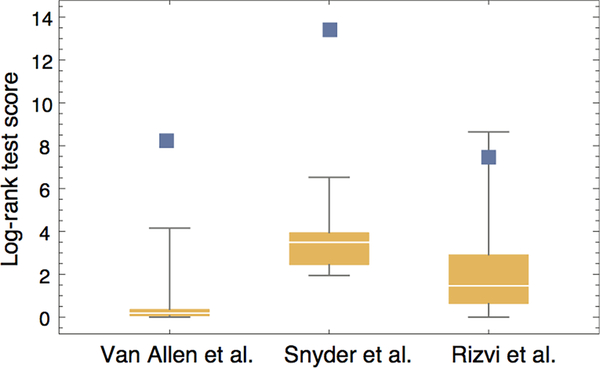
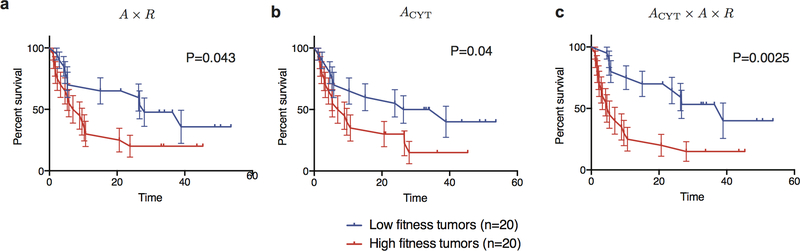
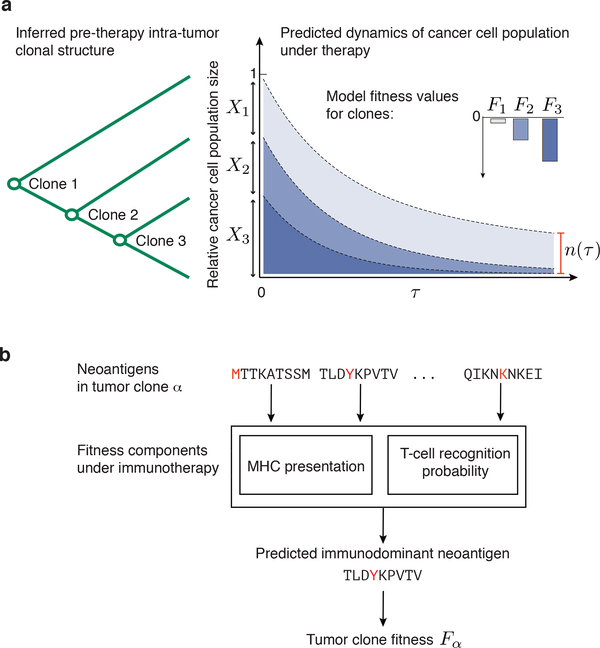
Comment in
-
How T cells spot tumour cells.Nature. 2017 Nov 23;551(7681):444-446. doi: 10.1038/d41586-017-07267-9. Nature. 2017. PMID: 29168843 No abstract available.
-
Immunotherapy: Relying on quality over quantity.Nat Rev Clin Oncol. 2018 Jan;15(1):6-7. doi: 10.1038/nrclinonc.2017.189. Epub 2017 Nov 28. Nat Rev Clin Oncol. 2018. PMID: 29182163 No abstract available.
References
-
- Schumacher TN & Schreiber RD Neoantigens in cancer immunotherapy. Science 348, 69–74 (2015). - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
