

Decreased ceramide underlies mitochondrial dysfunction in Charcot-Marie-Tooth 2F
- PMID: 29133339
- PMCID: PMC5892732
- DOI: 10.1096/fj.201701067R
Decreased ceramide underlies mitochondrial dysfunction in Charcot-Marie-Tooth 2F
Abstract
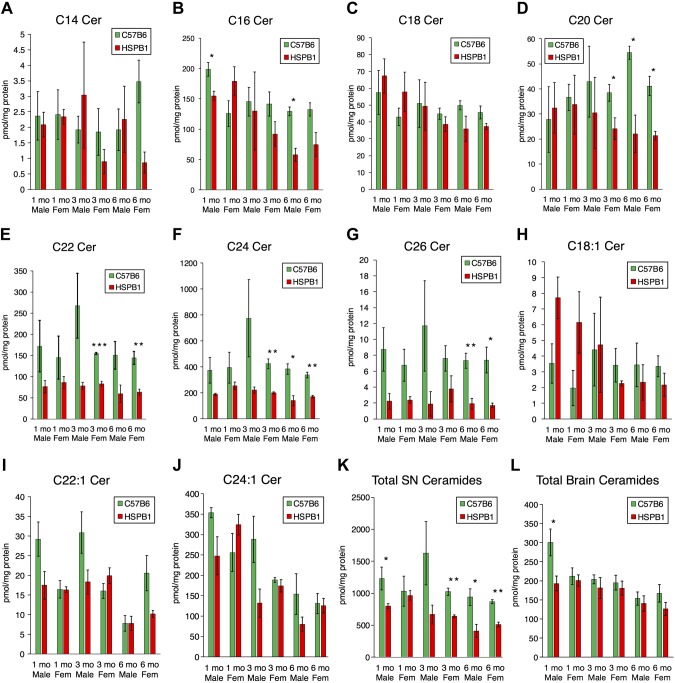
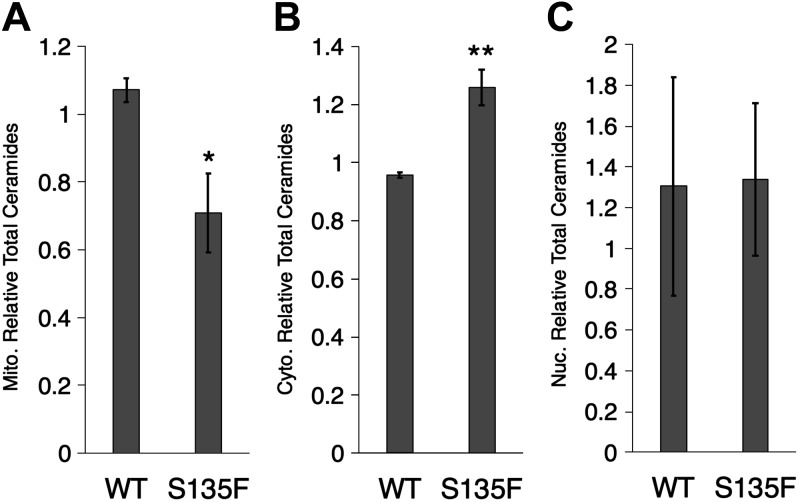
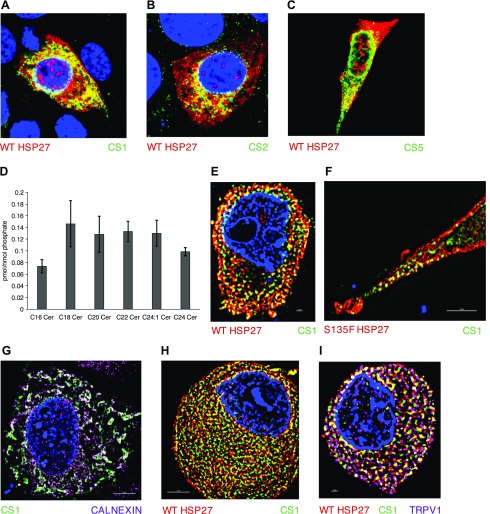
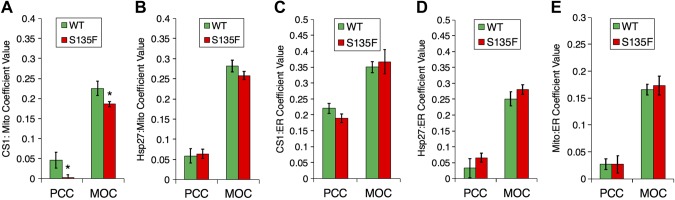
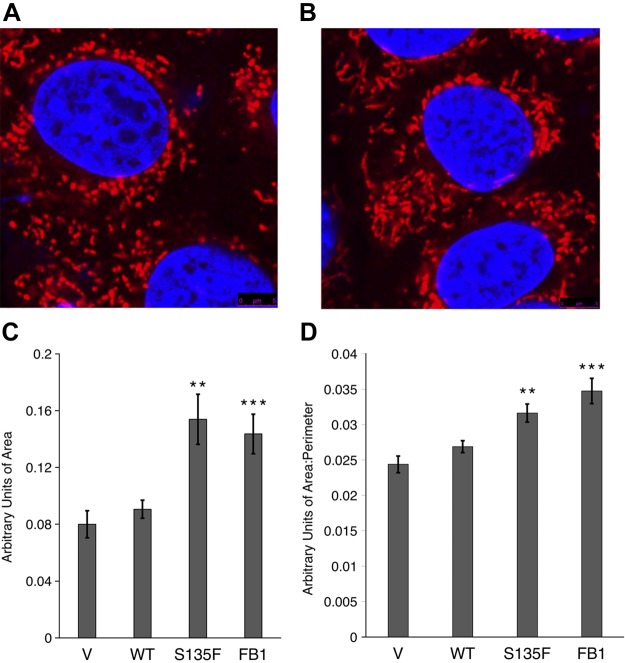
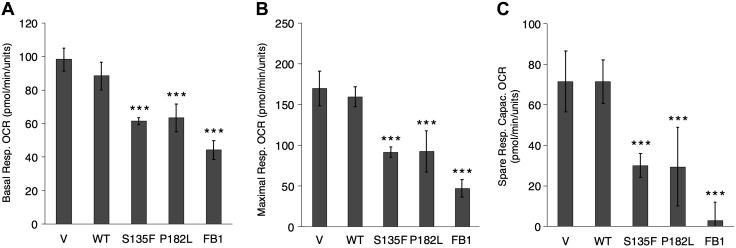
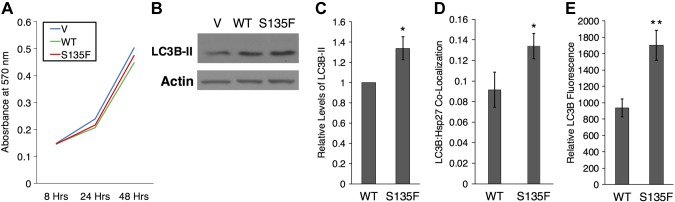
Charcot-Marie-Tooth (CMT) disease is the most commonly inherited neurologic disorder, but its molecular mechanisms remain unclear. One variant of CMT, 2F, is characterized by mutations in heat shock protein 27 (Hsp27). As bioactive sphingolipids have been implicated in neurodegenerative diseases, we sought to determine if their dysregulation is involved in CMT. Here, we show that Hsp27 knockout mice demonstrated decreases in ceramide in peripheral nerve tissue and that the disease-associated Hsp27 S135F mutant demonstrated decreases in mitochondrial ceramide. Given that Hsp27 is a chaperone protein, we examined its role in regulating ceramide synthases (CerSs), an enzyme family responsible for catalyzing generation of the sphingolipid ceramide. We determined that CerSs colocalized with Hsp27, and upon the presence of S135F mutants, CerS1 lost its colocalization with mitochondria suggesting that decreased mitochondrial ceramides result from reduced mitochondrial CerS localization rather than decreased CerS activity. Mitochondria in mutant cells appeared larger with increased interconnectivity. Furthermore, mutant cell lines demonstrated decreased mitochondrial respiratory function and increased autophagic flux. Mitochondrial structural and functional changes were recapitulated by blocking ceramide generation pharmacologically. These results suggest that mutant Hsp27 decreases mitochondrial ceramide levels, producing structural and functional changes in mitochondria leading to neuronal degeneration.-Schwartz, N. U., Linzer, R. W., Truman, J.-P., Gurevich, M., Hannun, Y. A., Senkal, C. E., Obeid, L. M. Decreased ceramide underlies mitochondrial dysfunction in Charcot-Marie-Tooth 2F.
Keywords: CMT2F; CerS; mitochondria; neuropathy; sphingolipid.
Conflict of interest statement
The authors thank Dr. Chiara Luberto (Stony Brook University) for overall advice. The authors also thank the Stony Brook Lipidomics Core for measurement and analysis of sphingolipids, the Stony Brook Proteomics Core for proteomic measurement and analysis of samples, and Dr. Ashley Snider (Stony Brook University) for assistance in managing mice. Peter Dong (University of Pennsylvania, Philadelphia, PA, USA) and Dr. Tanvir Khan (Stony Brook University) assisted in training for DRG dissection. The research in this manuscript was supported by the U.S. National Institutes of Health (NIH) National Institute of General Medical Sciences Grant GM062887 and Veterans Affairs Merit Award 5I01BX000156-08 (to L.M.O.), NIH National Cancer Institute Grant P01CA097132 (to Y.A.H. and L.M.O.). The authors declare no conflicts of interest.
Figures
References
-
- Timmerman V., Strickland A. V., Züchner S. (2014) Genetics of Charcot-Marie-Tooth (CMT) disease within the frame of the human genome project success. Genes (Basel) 5, 13–32https://doi.org/10.3390/genes5010013 - DOI - PMC - PubMed
-
- Dyck P. J., Lambert E. H. (1968) Lower motor and primary sensory neuron diseases with peroneal muscular atrophy. I. Neurologic, genetic, and electrophysiologic findings in hereditary polyneuropathies. Arch. Neurol. 18, 603–618https://doi.org/10.1001/archneur.1968.00470360025002 - DOI - PubMed
-
- MacMillan J. C., Harper P. S. (1994) The Charcot-Marie-Tooth syndrome: clinical aspects from a population study in South Wales, UK. Clin. Genet. 45, 128–134https://doi.org/10.1111/j.1399-0004.1994.tb04009.x - DOI - PubMed
-
- Harel T., Lupski J. R. (2014) Charcot-Marie-Tooth disease and pathways to molecular based therapies. Clin. Genet. 86, 422–431https://doi.org/10.1111/cge.12393 - DOI - PubMed
-
- Evgrafov O. V., Mersiyanova I., Irobi J., Van Den Bosch L., Dierick I., Leung C. L., Schagina O., Verpoorten N., Van Impe K., Fedotov V., Dadali E., Auer-Grumbach M., Windpassinger C., Wagner K., Mitrovic Z., Hilton-Jones D., Talbot K., Martin J.-J., Vasserman N., Tverskaya S., Polyakov A., Liem R. K. H., Gettemans J., Robberecht W., De Jonghe P., Timmerman V. (2004) Mutant small heat-shock protein 27 causes axonal Charcot-Marie-Tooth disease and distal hereditary motor neuropathy. Nat. Genet. 36, 602–606https://doi.org/10.1038/ng1354 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
