

Specific inhibition of GPCR-independent G protein signaling by a rationally engineered protein
- PMID: 29133411
- PMCID: PMC5715746
- DOI: 10.1073/pnas.1707992114
Specific inhibition of GPCR-independent G protein signaling by a rationally engineered protein
Abstract
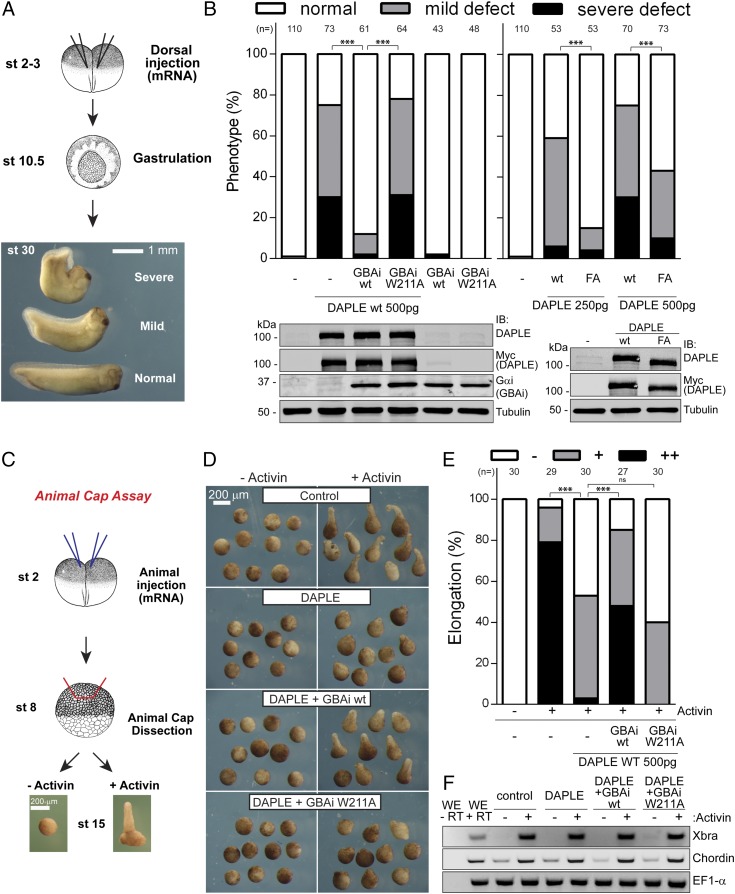
Activation of heterotrimeric G proteins by cytoplasmic nonreceptor proteins is an alternative to the classical mechanism via G protein-coupled receptors (GPCRs). A subset of nonreceptor G protein activators is characterized by a conserved sequence named the Gα-binding and activating (GBA) motif, which confers guanine nucleotide exchange factor (GEF) activity in vitro and promotes G protein-dependent signaling in cells. GBA proteins have important roles in physiology and disease but remain greatly understudied. This is due, in part, to the lack of efficient tools that specifically disrupt GBA motif function in the context of the large multifunctional proteins in which they are embedded. This hindrance to the study of alternative mechanisms of G protein activation contrasts with the wealth of convenient chemical and genetic tools to manipulate GPCR-dependent activation. Here, we describe the rational design and implementation of a genetically encoded protein that specifically inhibits GBA motifs: GBA inhibitor (GBAi). GBAi was engineered by introducing modifications in Gαi that preclude coupling to every known major binding partner [GPCRs, Gβγ, effectors, guanine nucleotide dissociation inhibitors (GDIs), GTPase-activating proteins (GAPs), or the chaperone/GEF Ric-8A], while favoring high-affinity binding to all known GBA motifs. We demonstrate that GBAi does not interfere with canonical GPCR-G protein signaling but blocks GBA-dependent signaling in cancer cells. Furthermore, by implementing GBAi in vivo, we show that GBA-dependent signaling modulates phenotypes during Xenopus laevis embryonic development. In summary, GBAi is a selective, efficient, and convenient tool to dissect the biological processes controlled by a GPCR-independent mechanism of G protein activation mediated by cytoplasmic factors.
Keywords: DAPLE; GEF; GPCR; Girdin; integrin.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
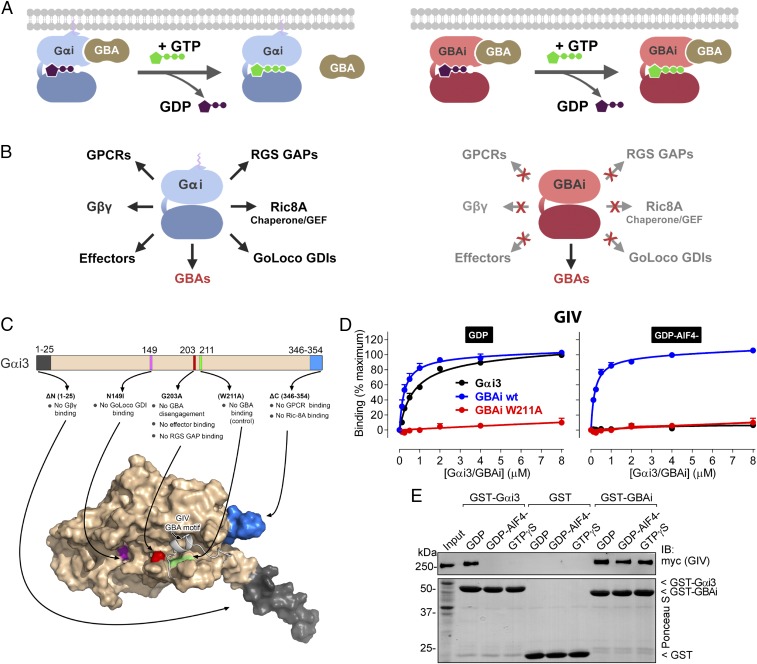
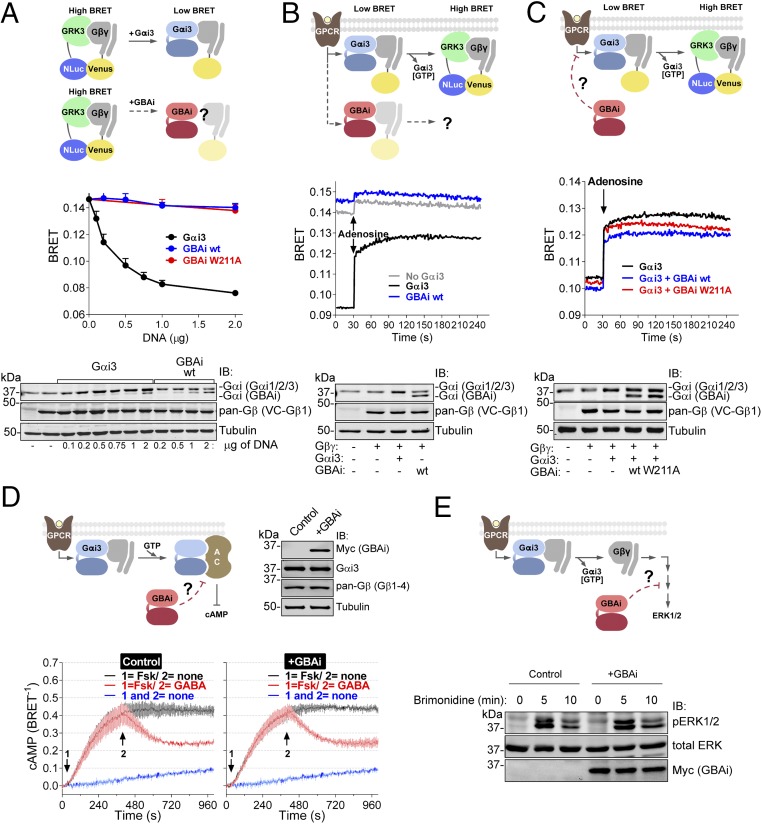
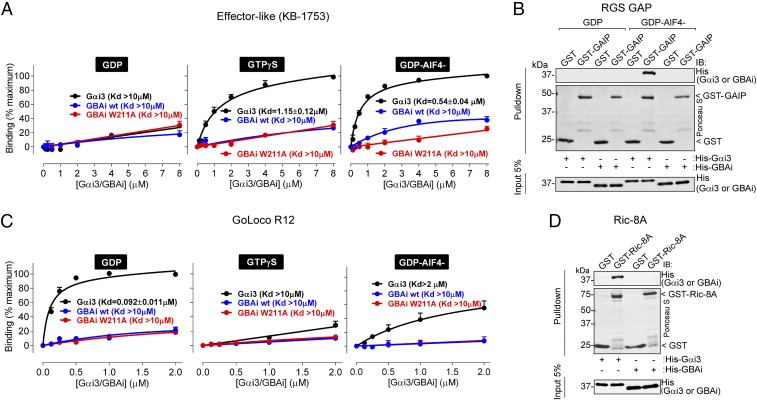
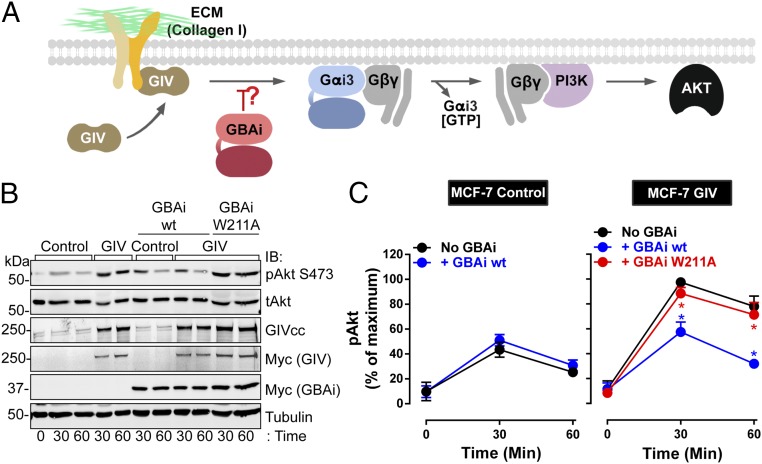
References
-
- Gilman AG. G proteins: Transducers of receptor-generated signals. Annu Rev Biochem. 1987;56:615–649. - PubMed
-
- Sato M, Blumer JB, Simon V, Lanier SM. Accessory proteins for G proteins: Partners in signaling. Annu Rev Pharmacol Toxicol. 2006;46:151–187. - PubMed
-
- Willard FS, Kimple RJ, Siderovski DP. Return of the GDI: The GoLoco motif in cell division. Annu Rev Biochem. 2004;73:925–951. - PubMed
-
- Ross EM, Wilkie TM. GTPase-activating proteins for heterotrimeric G proteins: Regulators of G protein signaling (RGS) and RGS-like proteins. Annu Rev Biochem. 2000;69:795–827. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
