

Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells
- PMID: 29133788
- PMCID: PMC5684340
- DOI: 10.1038/s41467-017-01629-7
Regulation of angiotensin II actions by enhancers and super-enhancers in vascular smooth muscle cells
Abstract
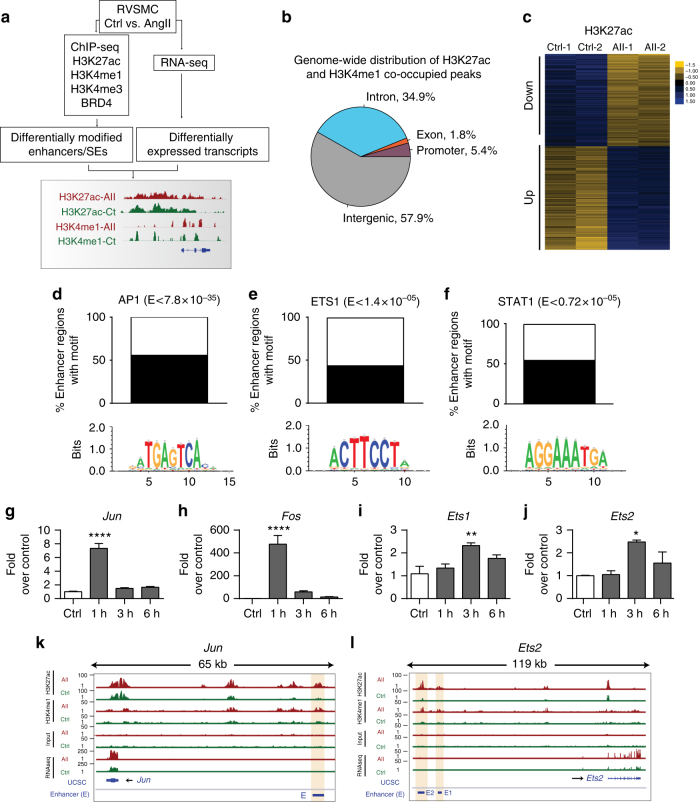
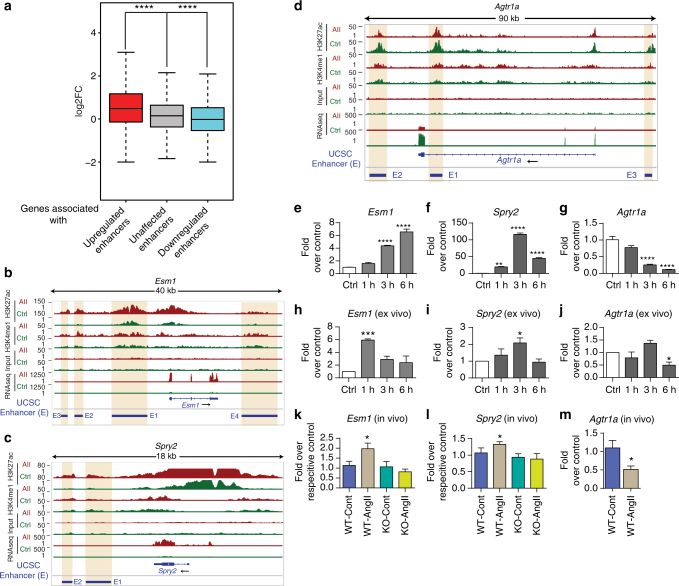
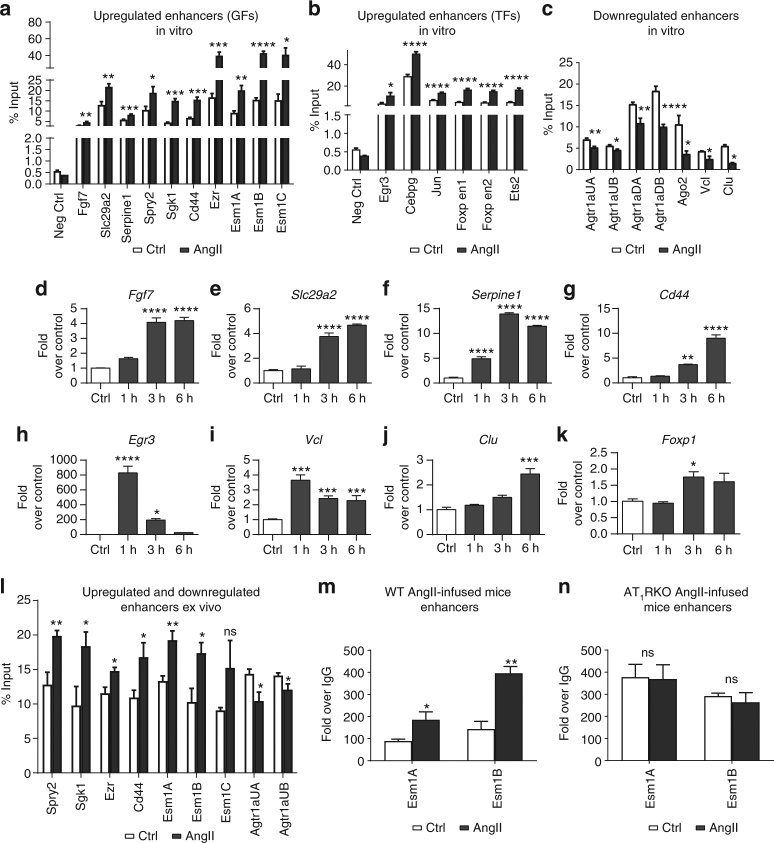
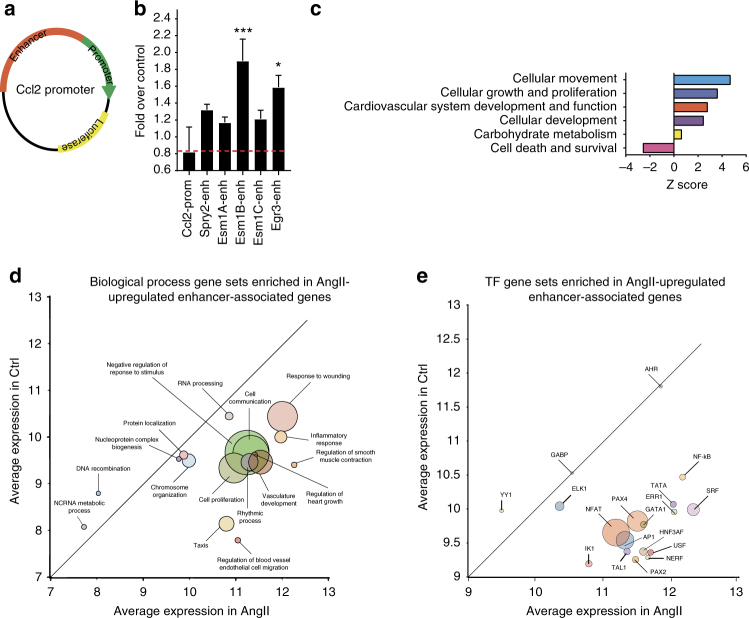
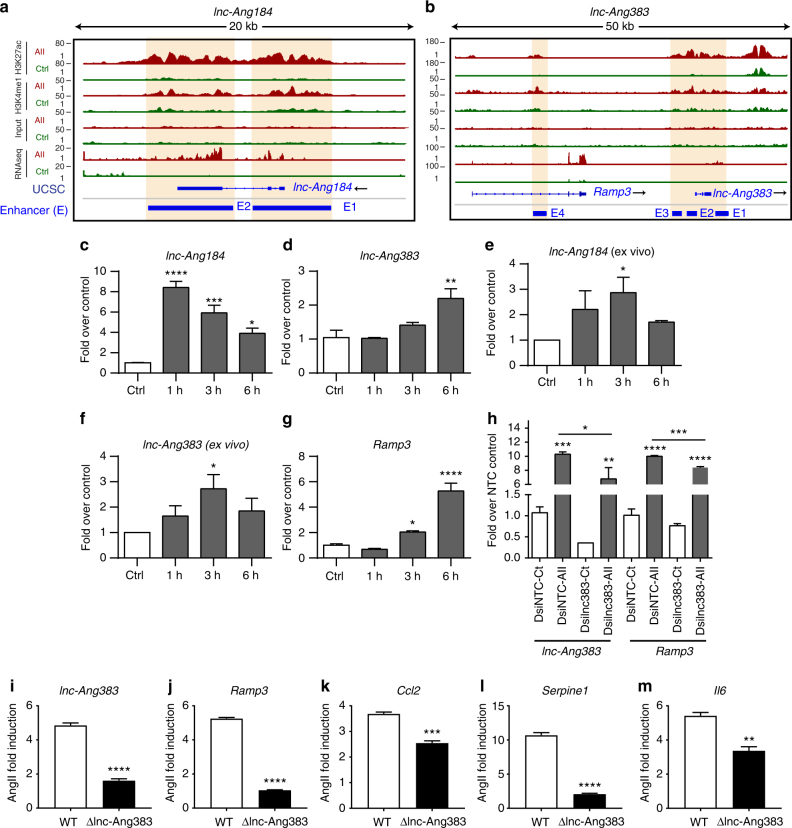
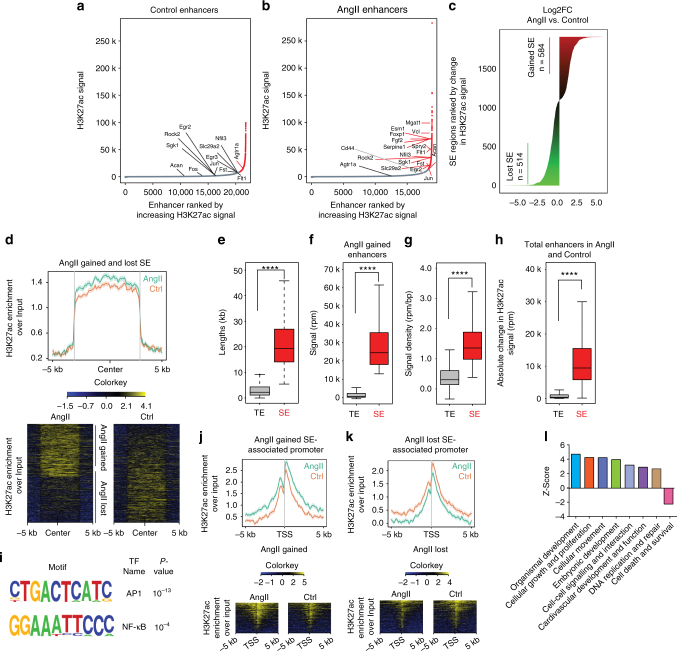
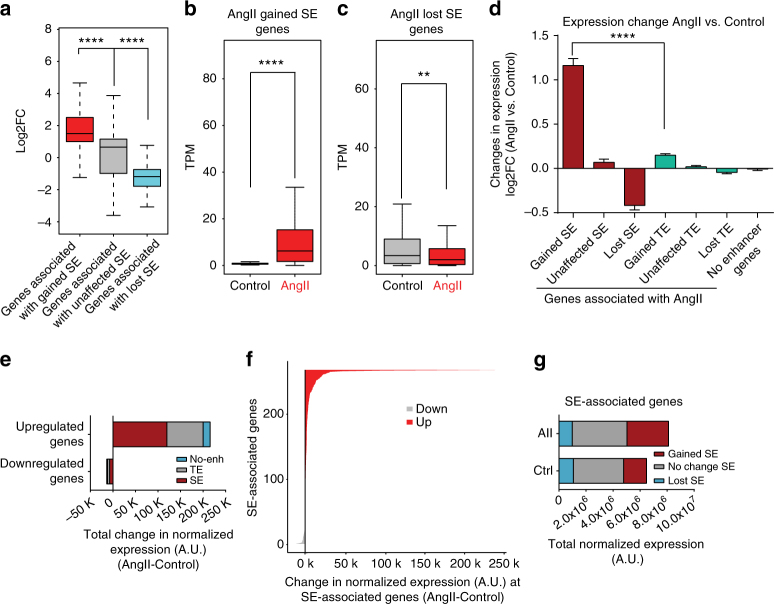
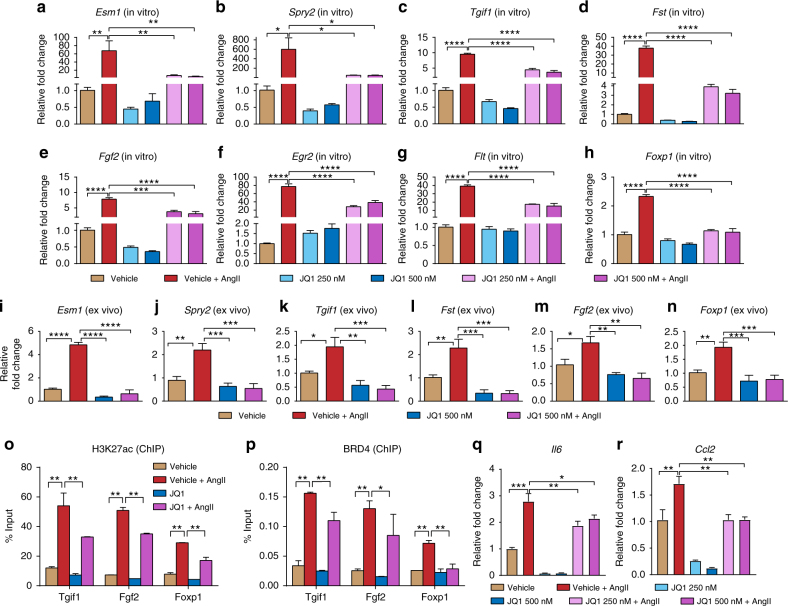
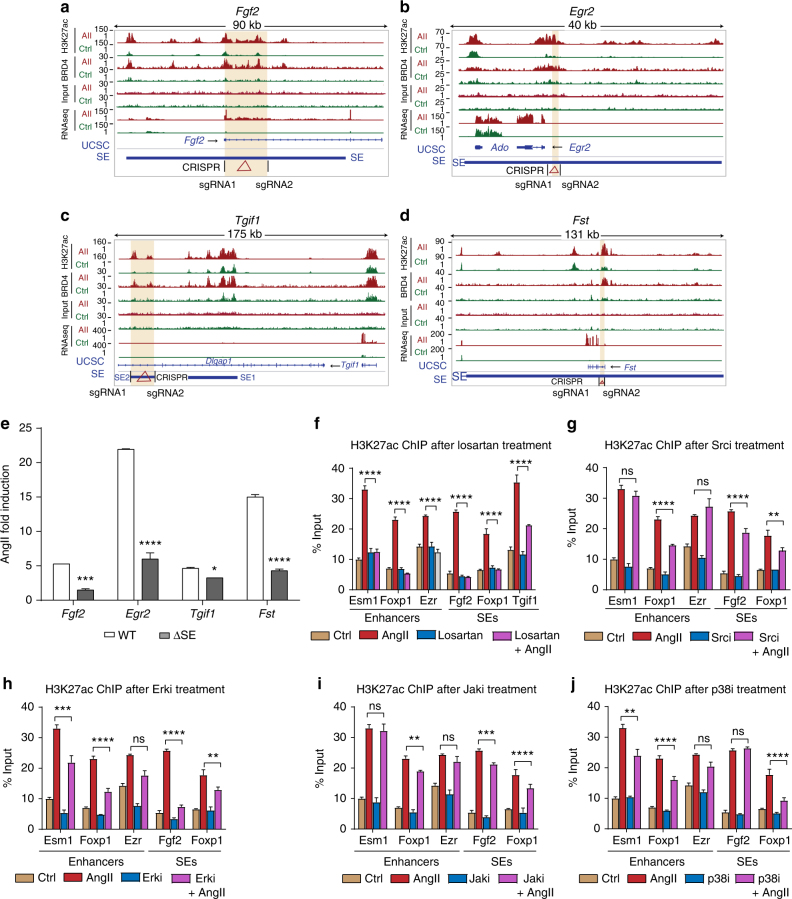
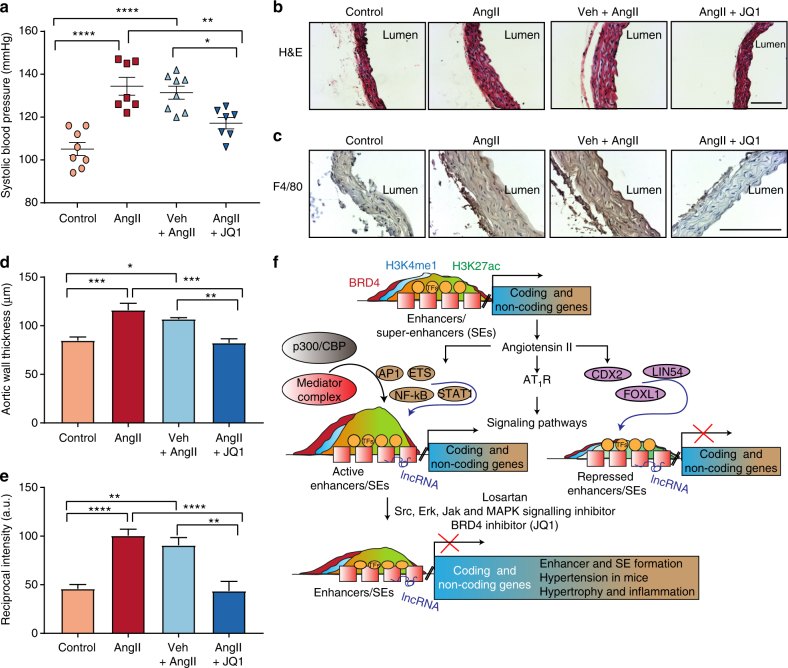
Angiotensin II (AngII) promotes hypertension and atherosclerosis by activating growth-promoting and pro-inflammatory gene expression in vascular smooth muscle cells (VSMCs). Enhancers and super-enhancers (SEs) play critical roles in driving disease-associated gene expression. However, enhancers/SEs mediating VSMC dysfunction remain uncharacterized. Here, we show that AngII alters vascular enhancer and SE repertoires in cultured VSMCs in vitro, ex vivo, and in AngII-infused mice aortas in vivo. AngII-induced enhancers/SEs are enriched in binding sites for signal-dependent transcription factors and dependent on key signaling kinases. Moreover, CRISPR-Cas9-mediated deletion of candidate enhancers/SEs, targeting SEs with the bromodomain and extra-terminal domain inhibitor JQ1, or knockdown of overlapping long noncoding RNAs (lncRNAs) blocks AngII-induced genes associated with growth-factor signaling and atherosclerosis. Furthermore, JQ1 ameliorates AngII-induced hypertension, medial hypertrophy and inflammation in vivo in mice. These results demonstrate AngII-induced signals integrate enhancers/SEs and lncRNAs to increase expression of genes involved in VSMC dysfunction, and could uncover novel therapies.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
