

LinkedOmics: analyzing multi-omics data within and across 32 cancer types
- PMID: 29136207
- PMCID: PMC5753188
- DOI: 10.1093/nar/gkx1090
LinkedOmics: analyzing multi-omics data within and across 32 cancer types
Abstract
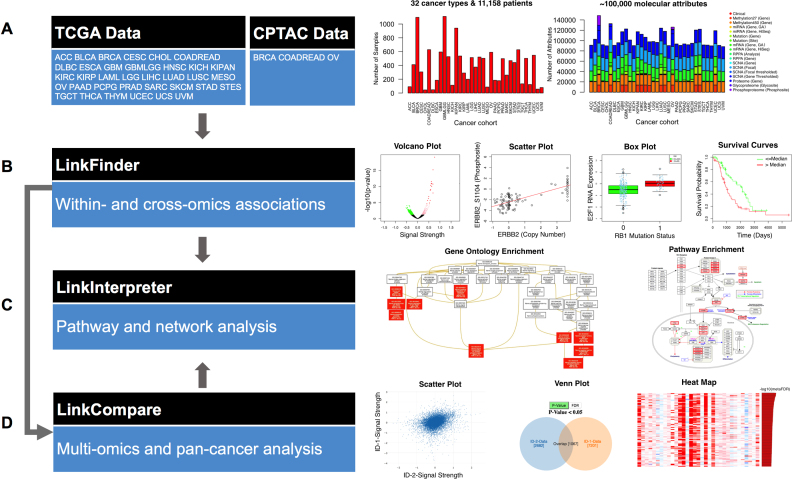
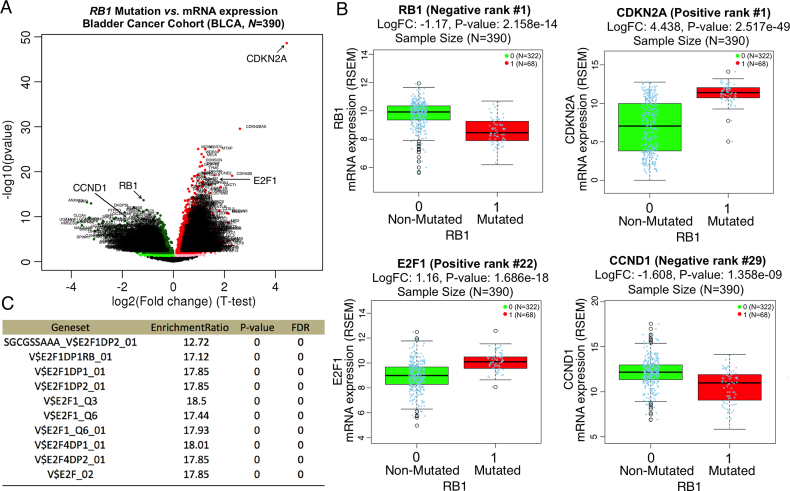
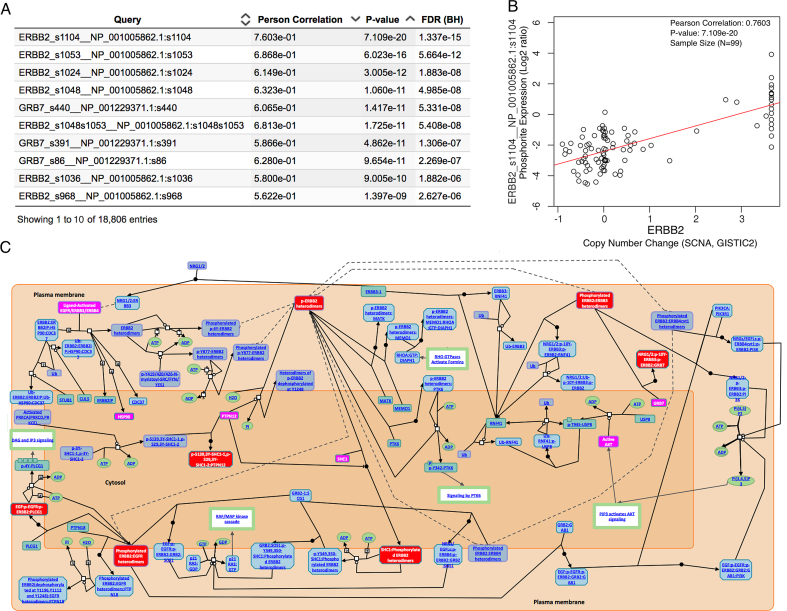
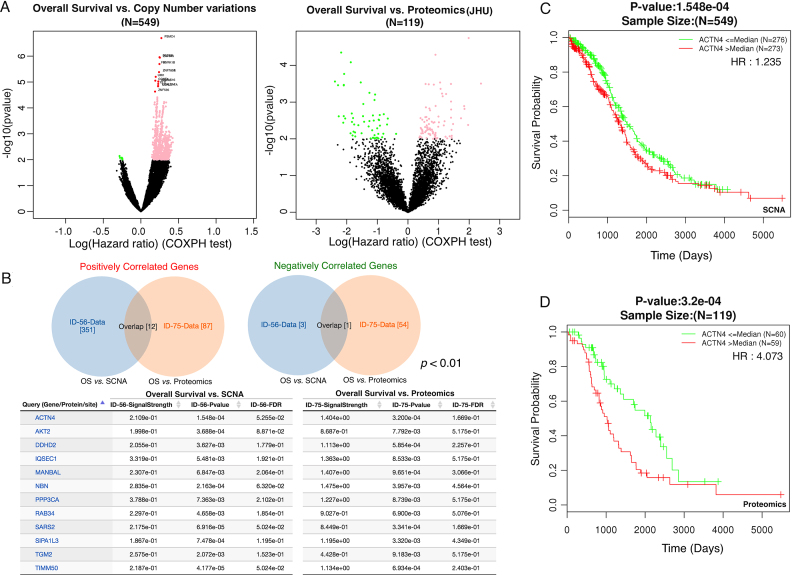
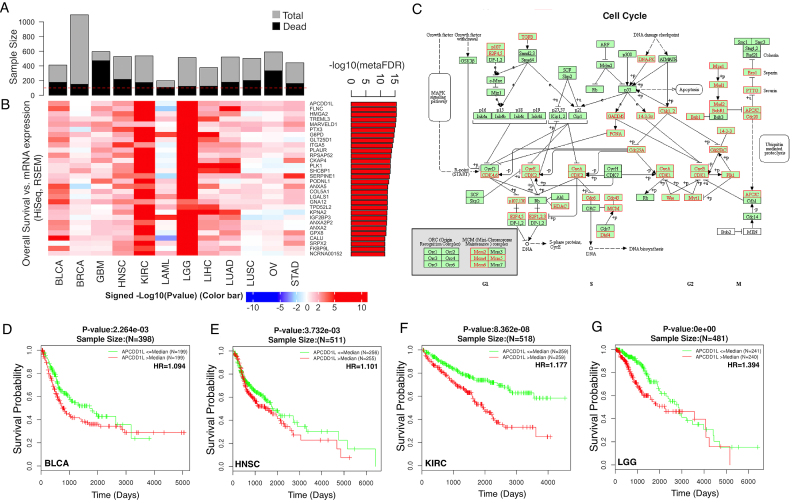
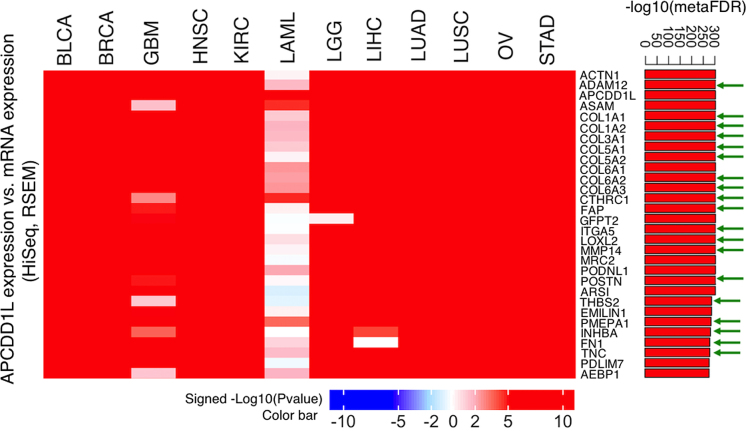
The LinkedOmics database contains multi-omics data and clinical data for 32 cancer types and a total of 11 158 patients from The Cancer Genome Atlas (TCGA) project. It is also the first multi-omics database that integrates mass spectrometry (MS)-based global proteomics data generated by the Clinical Proteomic Tumor Analysis Consortium (CPTAC) on selected TCGA tumor samples. In total, LinkedOmics has more than a billion data points. To allow comprehensive analysis of these data, we developed three analysis modules in the LinkedOmics web application. The LinkFinder module allows flexible exploration of associations between a molecular or clinical attribute of interest and all other attributes, providing the opportunity to analyze and visualize associations between billions of attribute pairs for each cancer cohort. The LinkCompare module enables easy comparison of the associations identified by LinkFinder, which is particularly useful in multi-omics and pan-cancer analyses. The LinkInterpreter module transforms identified associations into biological understanding through pathway and network analysis. Using five case studies, we demonstrate that LinkedOmics provides a unique platform for biologists and clinicians to access, analyze and compare cancer multi-omics data within and across tumor types. LinkedOmics is freely available at http://www.linkedomics.org.
© The Author(s) 2017. Published by Oxford University Press on behalf of Nucleic Acids Research.
Figures
References
-
- Fernandez-Banet J., Esposito A., Coffin S., Horvath I.B., Estrella H., Schefzick S., Deng S., Wang K., Aching K., Ding Y. et al. OASIS: web-based platform for exploring cancer multi-omics data. Nat. Methods. 2016; 13:9–10. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
