

Equilibrating (L)FeIII-OOAc and (L)FeV(O) Species in Hydrocarbon Oxidations by Bio-Inspired Nonheme Iron Catalysts Using H2O2 and AcOH
- PMID: 29136467
- PMCID: PMC5768304
- DOI: 10.1021/jacs.7b06246
Equilibrating (L)FeIII-OOAc and (L)FeV(O) Species in Hydrocarbon Oxidations by Bio-Inspired Nonheme Iron Catalysts Using H2O2 and AcOH
Abstract
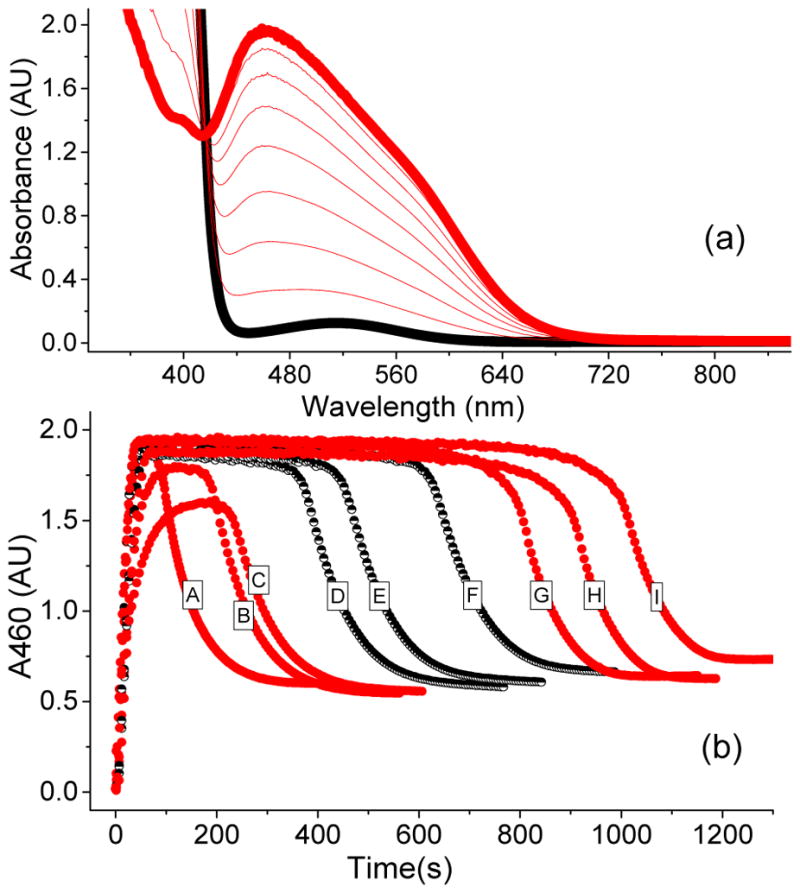
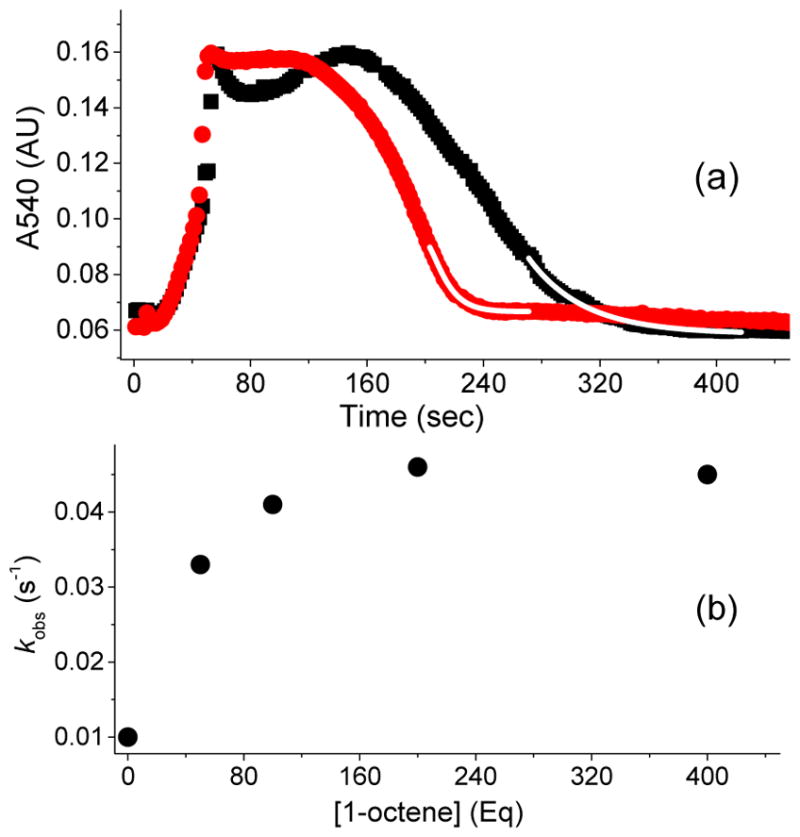
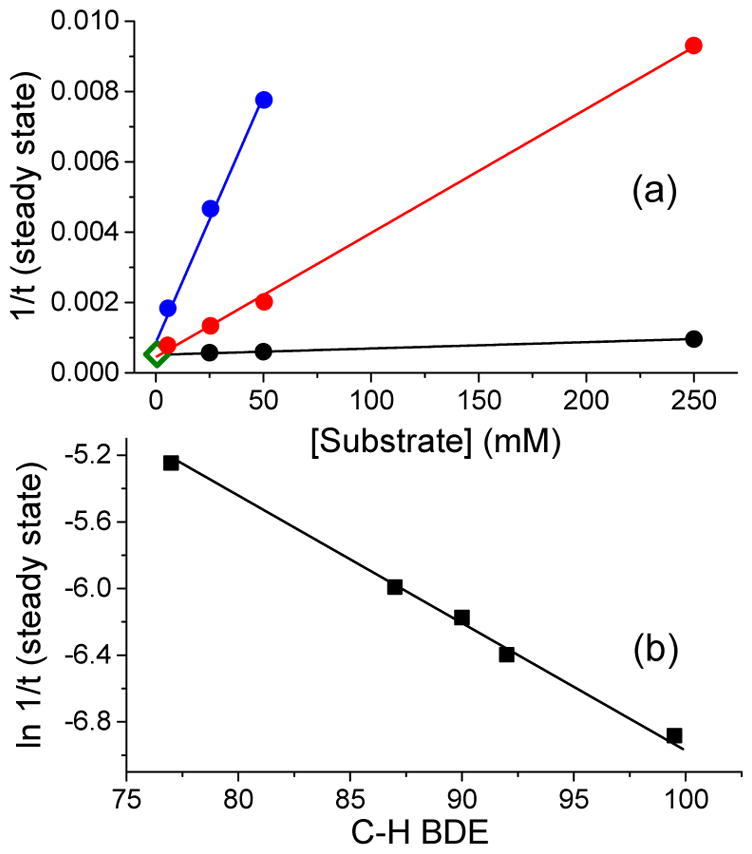
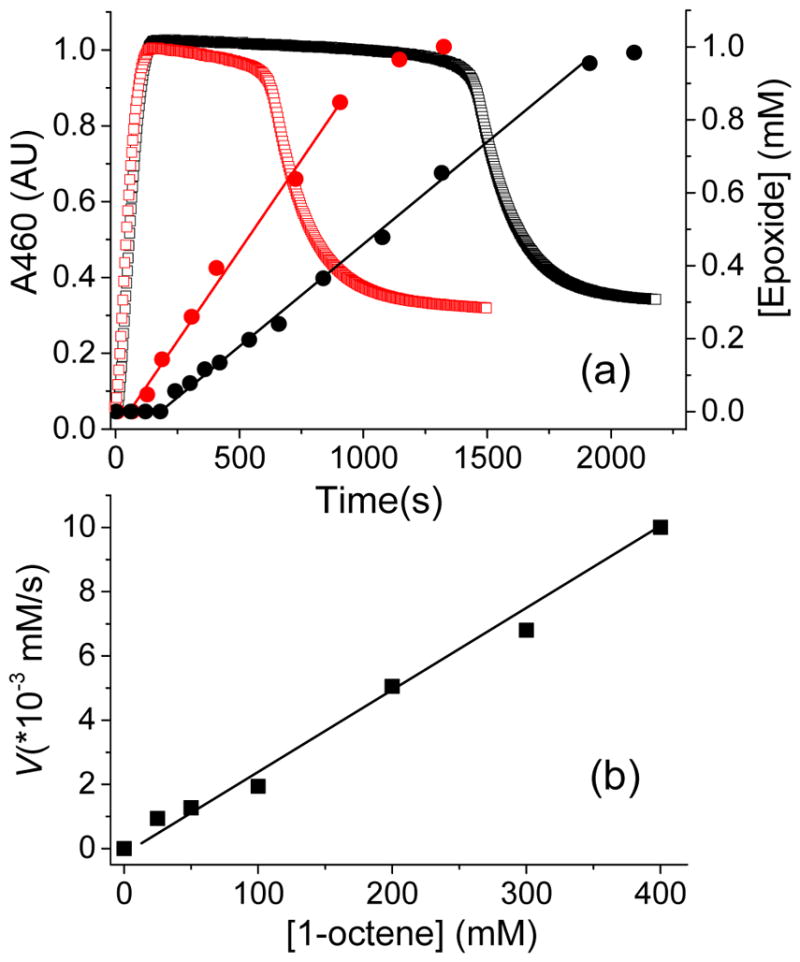
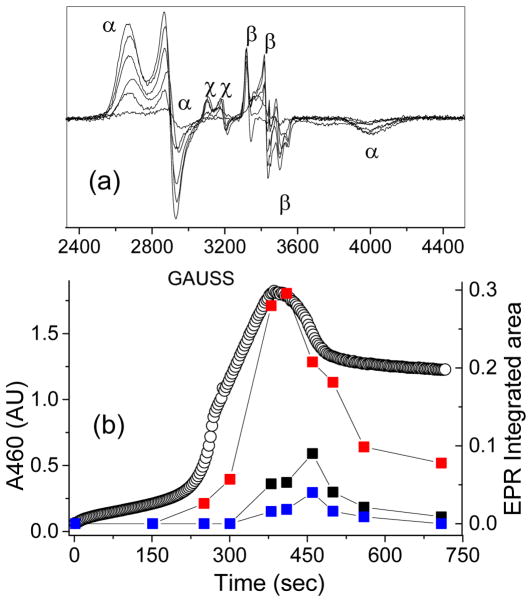
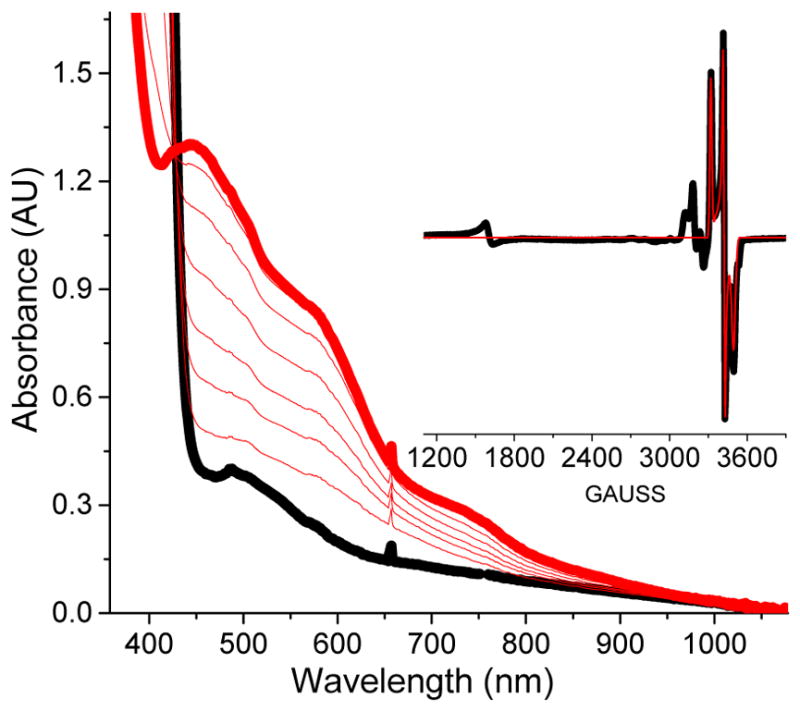
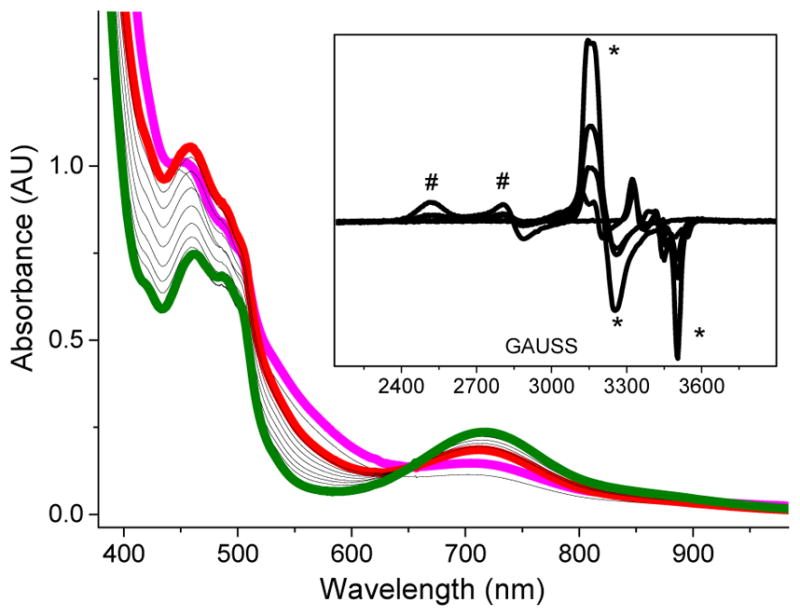
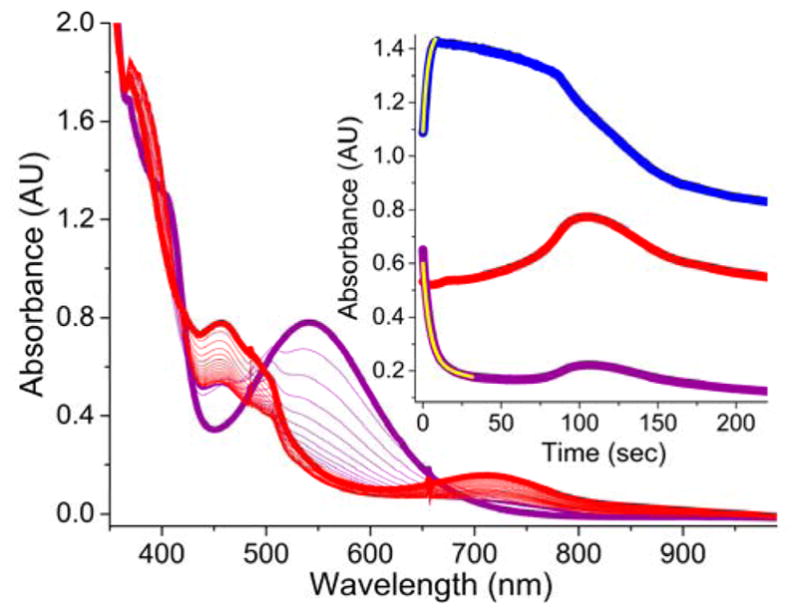
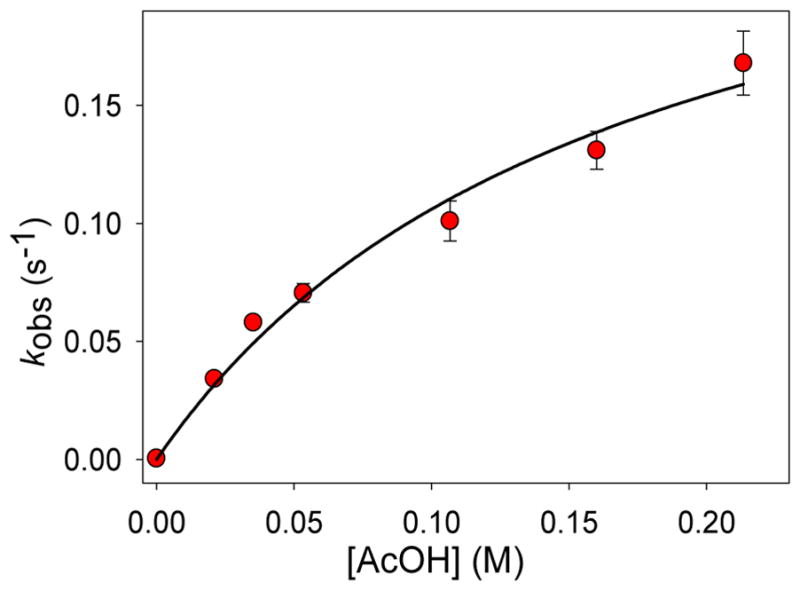
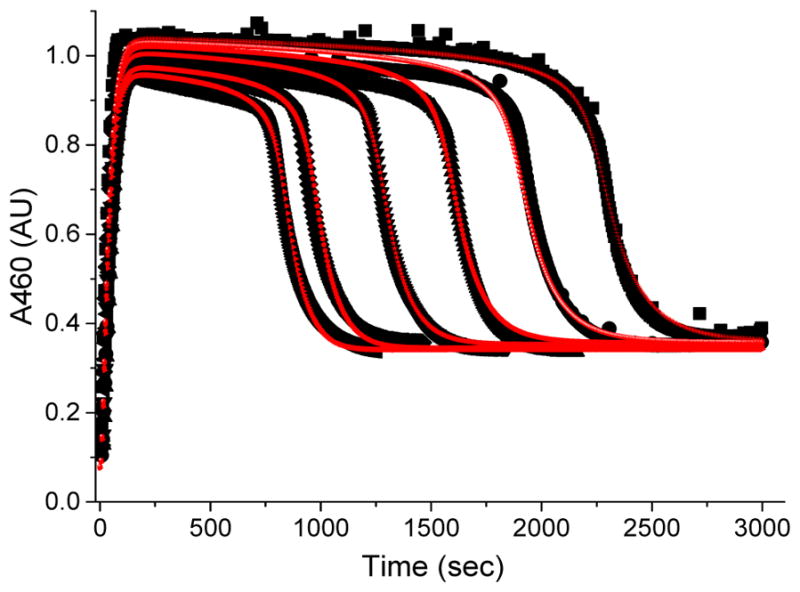
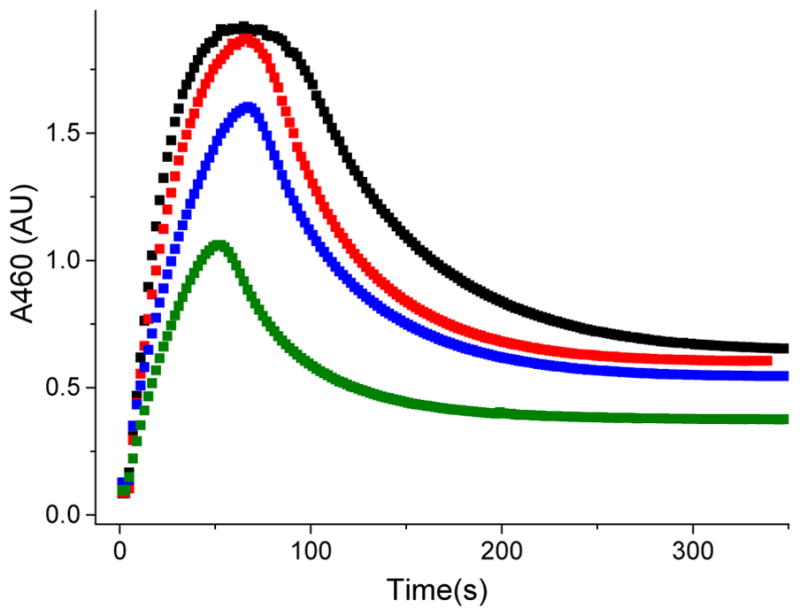
Inspired by the remarkable chemistry of the family of Rieske oxygenase enzymes, nonheme iron complexes of tetradentate N4 ligands have been developed to catalyze hydrocarbon oxidation reactions using H2O2 in the presence of added carboxylic acids. The observation that the stereo- and enantioselectivity of the oxidation products can be modulated by the electronic and steric properties of the acid implicates an oxidizing species that incorporates the carboxylate moiety. Frozen solutions of these catalytic mixtures generally exhibit EPR signals arising from two S = 1/2 intermediates, a highly anisotropic g2.7 subset (gmax = 2.58 to 2.78 and Δg = 0.85-1.2) that we assign to an FeIII-OOAc species and a less anisotropic g2.07 subset (g = 2.07, 2.01, and 1.96 and Δg ≈ 0.11) we associate with an FeV(O)(OAc) species. Kinetic studies on the reactions of iron complexes supported by the TPA (tris(pyridyl-2-methyl)amine) ligand family with H2O2/AcOH or AcOOH at -40 °C reveal the formation of a visible chromophore at 460 nm, which persists in a steady state phase and then decays exponentially upon depletion of the peroxo oxidant with a rate constant that is substrate independent. Remarkably, the duration of this steady state phase can be modulated by the nature of the substrate and its concentration, which is a rarely observed phenomenon. A numerical simulation of this behavior as a function of substrate type and concentration affords a kinetic model in which the two S = 1/2 intermediates exist in a dynamic equilibrium that is modulated by the electronic properties of the supporting ligands. This notion is supported by EPR studies of the reaction mixtures. Importantly, these studies unambiguously show that the g2.07 species, and not the g2.7 species, is responsible for substrate oxidation in the (L)FeII/H2O2/AcOH catalytic system. Instead the g2.7 species appears to be off-pathway and serves as a reservoir for the g2.07 species. These findings will be helpful not only for the design of regio- and stereospecific nonheme iron oxidation catalysts but also for providing insight into the mechanisms of the remarkably versatile oxidants formed by nature's most potent oxygenases.
Conflict of interest statement
Notes
The authors declare no competing financial interest.
Figures
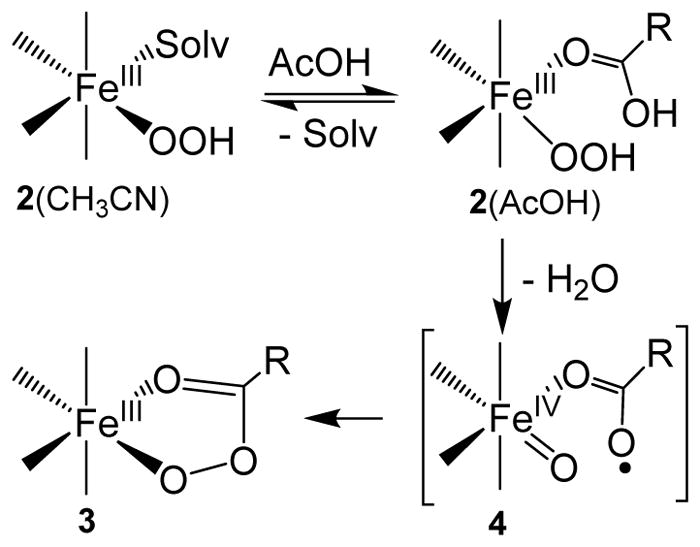
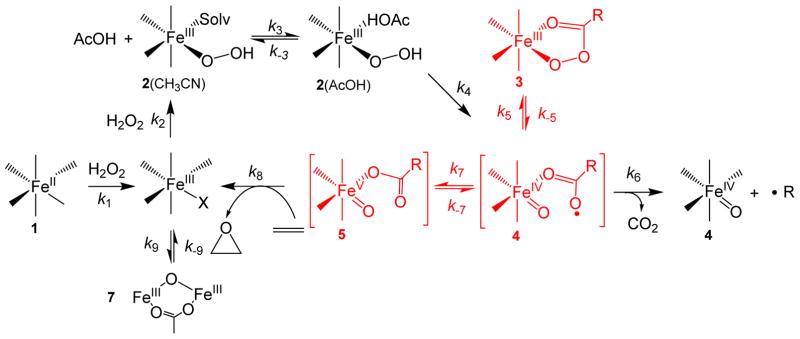
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
