

A novel polyamine blockade therapy activates an anti-tumor immune response
- PMID: 29137411
- PMCID: PMC5663583
- DOI: 10.18632/oncotarget.20493
A novel polyamine blockade therapy activates an anti-tumor immune response
Abstract
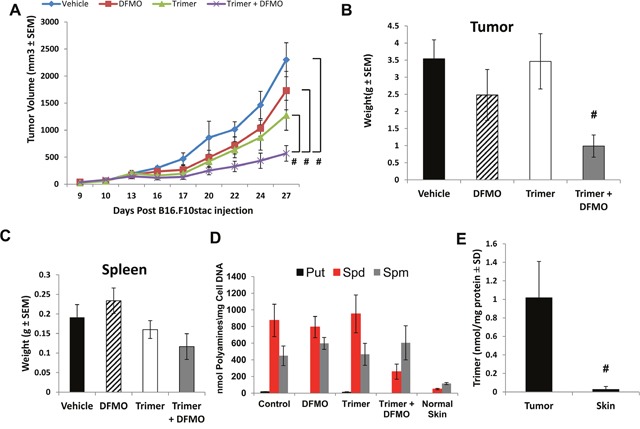
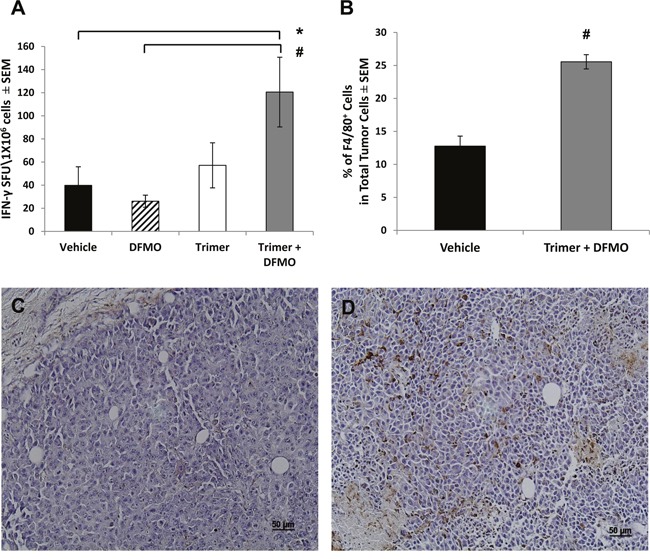
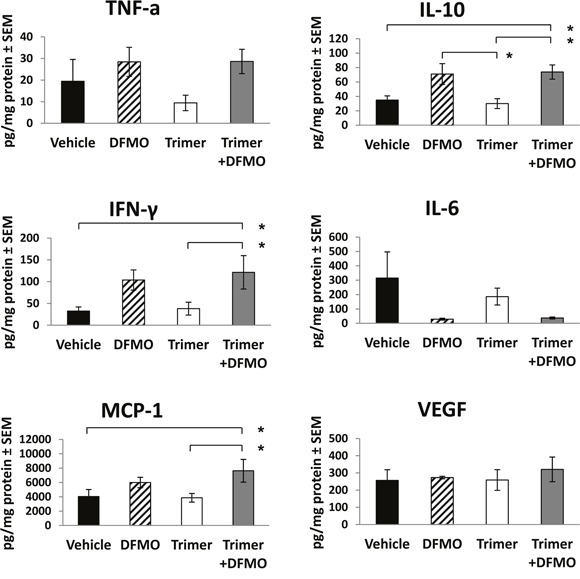
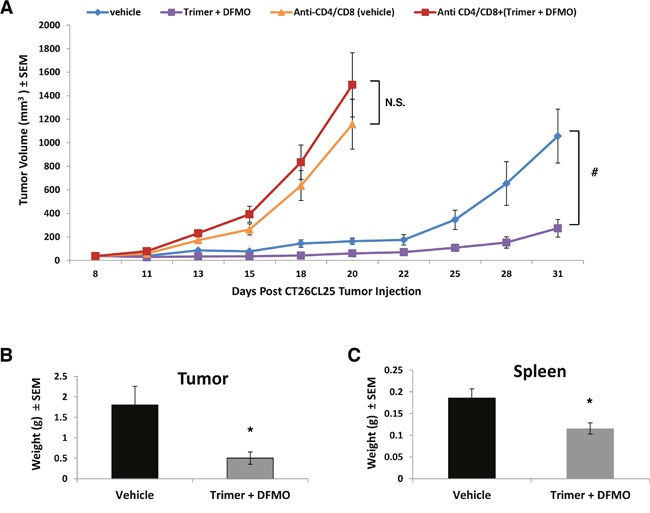
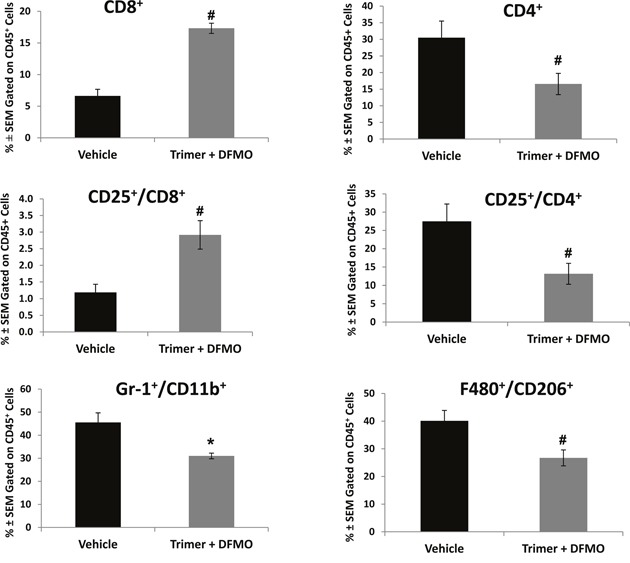
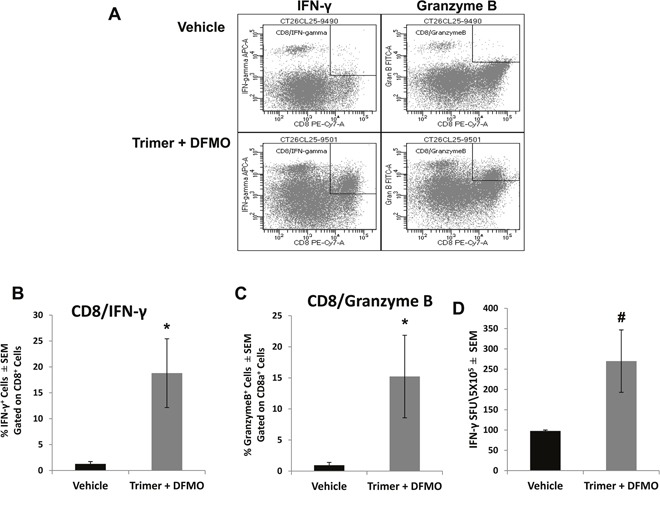
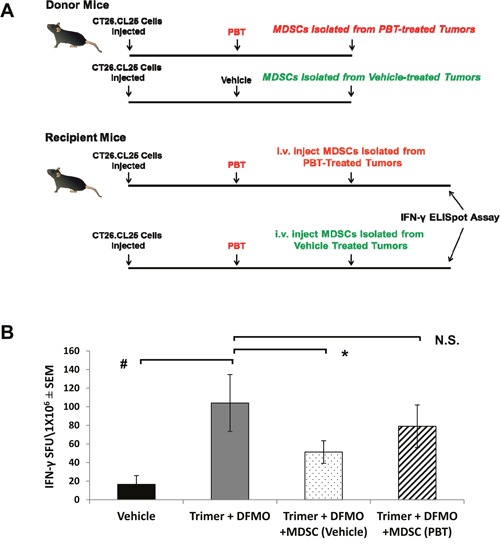
Most tumors maintain elevated levels of polyamines to support their growth and survival. This study explores the anti-tumor effect of polyamine starvation via both inhibiting polyamine biosynthesis and blocking the upregulated import of polyamines into the tumor. We demonstrate that polyamine blockade therapy (PBT) co-treatment with both DFMO and a novel polyamine transport inhibitor, Trimer PTI, significantly inhibits tumor growth more than treatment with DFMO or the Trimer PTI alone. The anti-tumor effect of PBT was lost in mice where CD4+ and CD8+ T cells were antibody depleted, implying that PBT stimulates an anti-tumor immune effect that is T-cell dependent. The PBT anti-tumor effect was accompanied by an increase in granzyme B+, IFN-γ+ CD8+ T-cells and a decrease in immunosuppressive tumor infiltrating cells including Gr-1+CD11b+ myeloid derived suppressor cells (MDSCs), CD4+CD25+ Tregs, and CD206+F4/80+ M2 macrophages. Stimulation with tumor-specific peptides elicited elevated antigen-specific IFN-γ secretion in splenocytes from PBT-treated mice, indicating that PBT treatment stimulates the activation of T-cells in a tumor-specific manner. These data show that combined treatment with both DFMO and the Trimer PTI not only deprives polyamine-addicted tumor cells of polyamines, but also relieves polyamine-mediated immunosuppression in the tumor microenvironment, thus allowing the activation of tumoricidal T-cells.
Keywords: difluoromethylornithine; immunomodulation; polyamines; transport inhibitor; tumor microenvironment.
Conflict of interest statement
CONFLICTS OF INTEREST Both the composition of matter and use of the Trimer PTI with DFMO in the treatment of cancers have been patented by UCF. As outlined in a collaborative research agreement, both UCF and the Lankenau Institute for Medical Research will equally share future royalties related to commercialization of this intellectual property. A patent application for the use of the Trimer PTI plus DFMO as immunomodulatory therapy has been filed jointly by LIMR and UCF.
Figures
Similar articles
-
Polyamine Blocking Therapy Decreases Survival of Tumor-Infiltrating Immunosuppressive Myeloid Cells and Enhances the Antitumor Efficacy of PD-1 Blockade.Mol Cancer Ther. 2020 Oct;19(10):2012-2022. doi: 10.1158/1535-7163.MCT-19-1116. Epub 2020 Aug 3. Mol Cancer Ther. 2020. PMID: 32747421 Free PMC article.
-
Loss of Anti-Tumor Efficacy by Polyamine Blocking Therapy in GCN2 Null Mice.Biomedicines. 2023 Oct 5;11(10):2703. doi: 10.3390/biomedicines11102703. Biomedicines. 2023. PMID: 37893077 Free PMC article.
-
Polyamine-blocking therapy reverses immunosuppression in the tumor microenvironment.Cancer Immunol Res. 2014 Mar;2(3):274-85. doi: 10.1158/2326-6066.CIR-13-0120-T. Epub 2013 Oct 7. Cancer Immunol Res. 2014. PMID: 24778323 Free PMC article.
-
Polyamine Depletion Strategies in Cancer: Remodeling the Tumor Immune Microenvironment to Enhance Anti-Tumor Responses.Med Sci (Basel). 2022 Jun 10;10(2):31. doi: 10.3390/medsci10020031. Med Sci (Basel). 2022. PMID: 35736351 Free PMC article. Review.
-
The role of polyamine metabolism in remodeling immune responses and blocking therapy within the tumor immune microenvironment.Front Immunol. 2022 Sep 2;13:912279. doi: 10.3389/fimmu.2022.912279. eCollection 2022. Front Immunol. 2022. PMID: 36119047 Free PMC article. Review.
Cited by
-
Therapeutic Targets in Diffuse Midline Gliomas-An Emerging Landscape.Cancers (Basel). 2021 Dec 13;13(24):6251. doi: 10.3390/cancers13246251. Cancers (Basel). 2021. PMID: 34944870 Free PMC article. Review.
-
Polyamines drive myeloid cell survival by buffering intracellular pH to promote immunosuppression in glioblastoma.Sci Adv. 2021 Feb 17;7(8):eabc8929. doi: 10.1126/sciadv.abc8929. Print 2021 Feb. Sci Adv. 2021. PMID: 33597238 Free PMC article.
-
Ornithine aminotransferase supports polyamine synthesis in pancreatic cancer.Nature. 2023 Apr;616(7956):339-347. doi: 10.1038/s41586-023-05891-2. Epub 2023 Mar 29. Nature. 2023. PMID: 36991126 Free PMC article.
-
Hyaluronate-coated perfluoroalkyl polyamine prodrugs as bioactive siRNA delivery systems for the treatment of peritoneal cancers.Biomater Adv. 2022 May;136:212755. doi: 10.1016/j.bioadv.2022.212755. Epub 2022 Mar 17. Biomater Adv. 2022. PMID: 35813988 Free PMC article.
-
Polyamines and related signaling pathways in cancer.Cancer Cell Int. 2020 Nov 5;20(1):539. doi: 10.1186/s12935-020-01545-9. Cancer Cell Int. 2020. PMID: 33292222 Free PMC article. Review.
References
-
- Rebucci M, Michiels C. Molecular aspects of cancer cell resistance to chemotherapy. Biochem Pharmacol. 2013;85:1219–26. https://doi.org/10.1016/j.bcp.2013.02.017. - DOI - PubMed
-
- Dobbelstein M, Moll U. Targeting tumour-supportive cellular machineries in anticancer drug development. Nat Rev Drug Discov. 2014;13:179–96. https://doi.org/10.1038/nrd4201. - DOI - PubMed
-
- Zhao J. Cancer stem cells and chemoresistance: the smartest survives the raid. Pharmacol Ther. 2016;160:145–58. https://doi.org/10.1016/j.pharmthera.2016.02.008. - DOI - PMC - PubMed
-
- Colotta F, Allavena P, Sica A, Garlanda C, Mantovani A. Cancer-related inflammation, the seventh hallmark of cancer: links to genetic instability. Carcinogenesis. 2009;30:1073–81. https://doi.org/10.1093/carcin/bgp127. - DOI - PubMed
-
- Pegg AE. Polyamine metabolism and its importance in neoplastic growth as a target for chemotherapy. Cancer Res. 1988;48:759–74. - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
