

Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer
- PMID: 29138515
- PMCID: PMC5686113
- DOI: 10.1038/s41598-017-15840-5
Targeting a non-oncogene addiction to the ATR/CHK1 axis for the treatment of small cell lung cancer
Abstract
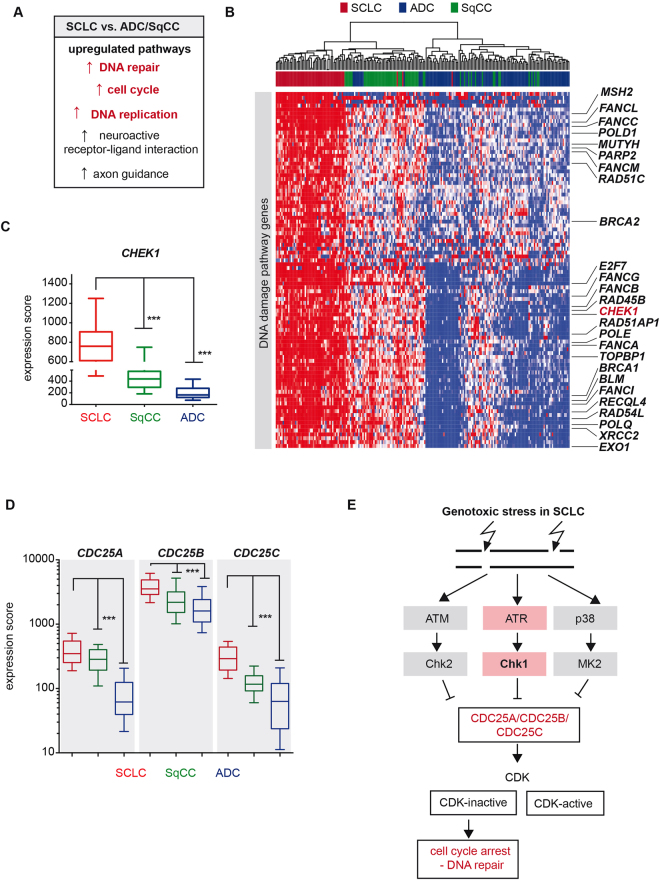
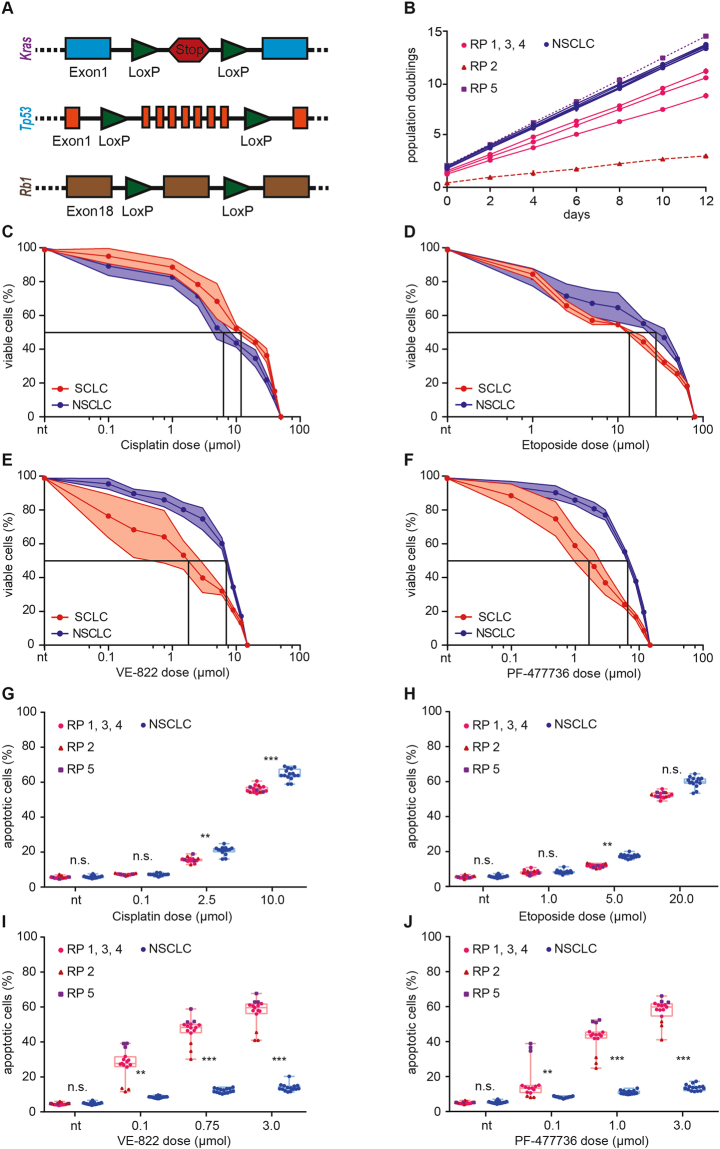
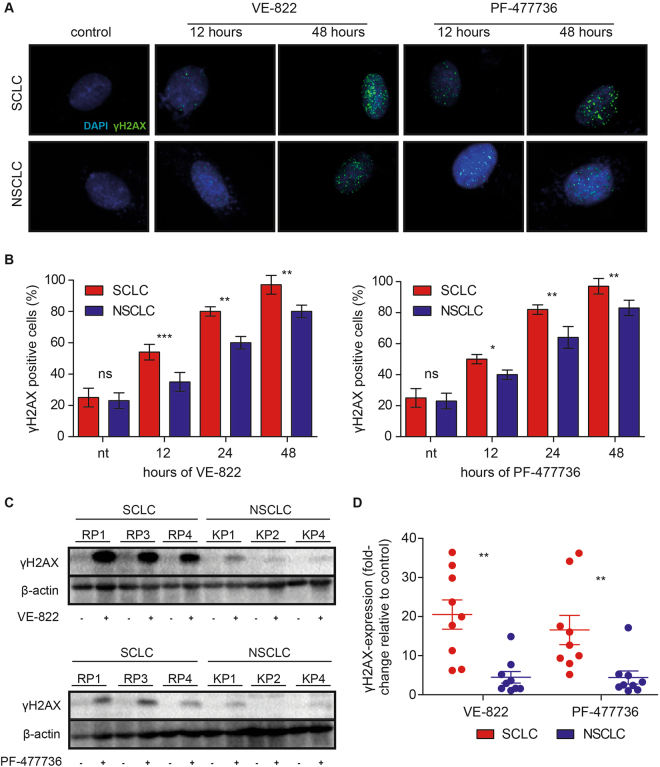
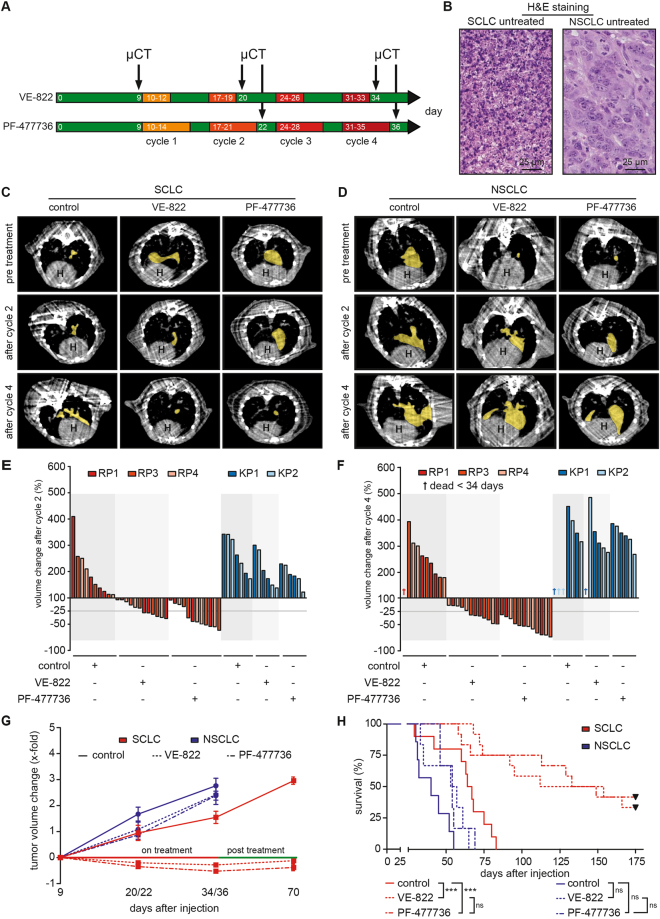
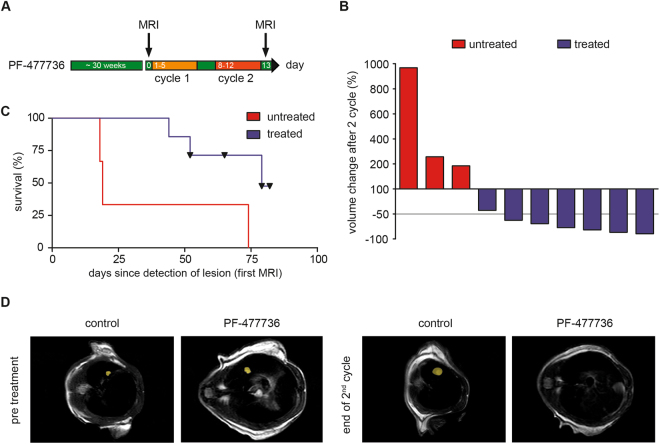
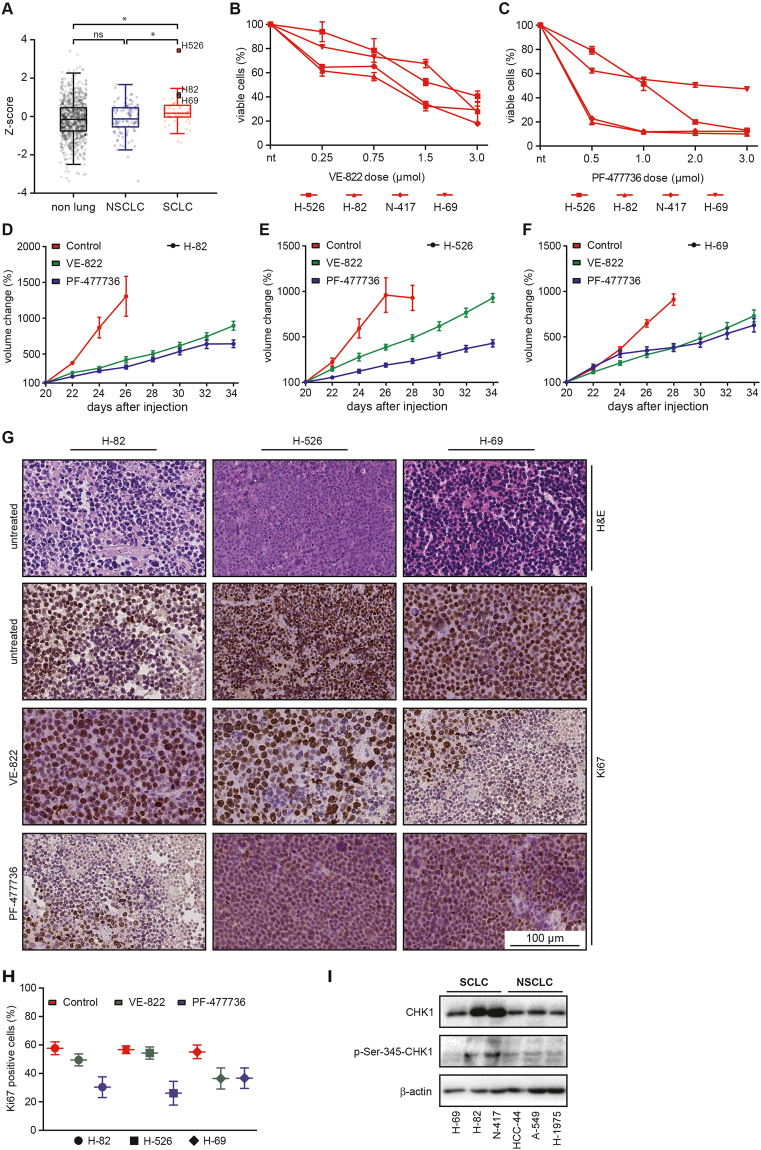
Small cell lung cancer (SCLC) is a difficult to treat subtype of lung cancer. One of the hallmarks of SCLC is its almost uniform chemotherapy sensitivity. However, chemotherapy response is typically transient and patients frequently succumb to SCLC within a year following diagnosis. We performed a transcriptome analysis of the major human lung cancer entities. We show a significant overexpression of genes involved in the DNA damage response, specifically in SCLC. Particularly CHEK1, which encodes for the cell cycle checkpoint kinase CHK1, is significantly overexpressed in SCLC, compared to lung adenocarcinoma. In line with uncontrolled cell cycle progression in SCLC, we find that CDC25A, B and C mRNAs are expressed at significantly higher levels in SCLC, compared to lung adenocarcinoma. We next profiled the efficacy of compounds targeting CHK1 and ATR. Both, ATR- and CHK1 inhibitors induce genotoxic damage and apoptosis in human and murine SCLC cell lines, but not in lung adenocarcinoma cells. We further demonstrate that murine SCLC tumors were highly sensitive to ATR- and CHK1 inhibitors, while Kras G12D -driven murine lung adenocarcinomas were resistant against these compounds and displayed continued growth under therapy. Altogether, our data indicate that SCLC displays an actionable dependence on ATR/CHK1-mediated cell cycle checkpoints.
Conflict of interest statement
H.C.R. received consulting and lecture fees from Abbvie, Astra-Zeneca, Vertex and Merck. R.K.T. is a founder of NEO New Oncology GmbH, now part of Siemens Healthcare. R.K.T. received consulting and lecture fees (Merck, Roche, Lilly, Boehringer Ingelheim, Astra-Zeneca, Daiichi-Sankyo, MSD, NEO New Oncology, Puma, Clovis). The remaining authors declare no competing financial interest.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
