

Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy
- PMID: 29142081
- PMCID: PMC5771312
- DOI: 10.1124/jpet.117.244673
Genetic and Molecular Regulation of Extrasynaptic GABA-A Receptors in the Brain: Therapeutic Insights for Epilepsy
Abstract
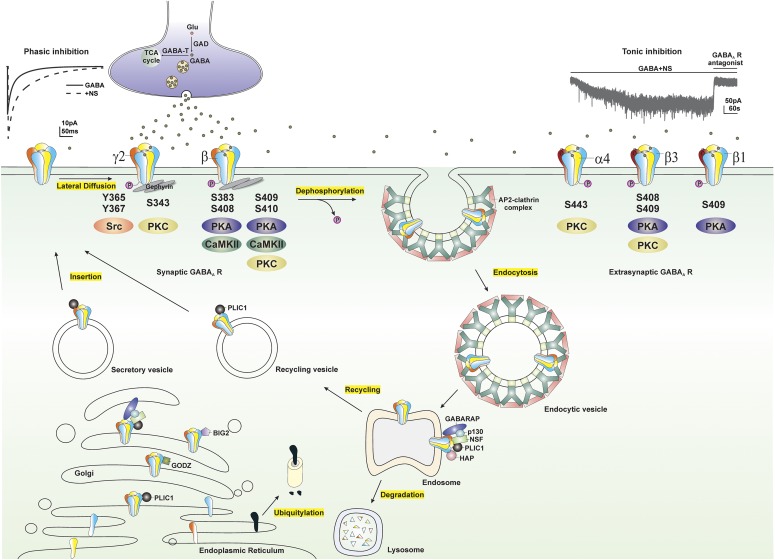
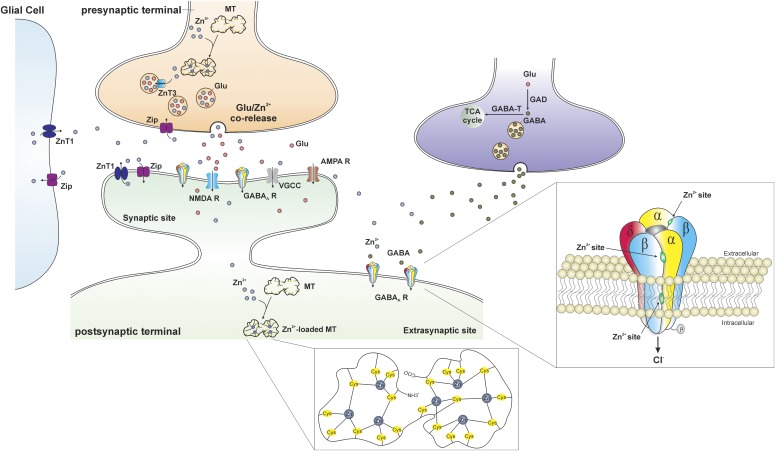
GABA-A receptors play a pivotal role in many brain diseases. Epilepsy is caused by acquired conditions and genetic defects in GABA receptor channels regulating neuronal excitability in the brain. The latter is referred to as GABA channelopathies. In the last two decades, major advances have been made in the genetics of epilepsy. The presence of specific GABAergic genetic abnormalities leading to some of the classic epileptic syndromes has been identified. Advances in molecular cloning and recombinant systems have helped characterize mutations in GABA-A receptor subunit genes in clinical neurology. GABA-A receptors are the prime targets for neurosteroids (NSs). However, GABA-A receptors are not static but undergo rapid changes in their number or composition in response to the neuroendocrine milieu. This review describes the recent advances in the genetic and neuroendocrine control of extrasynaptic and synaptic GABA-A receptors in epilepsy and its impact on neurologic conditions. It highlights the current knowledge of GABA genetics in epilepsy, with an emphasis on the neuroendocrine regulation of extrasynaptic GABA-A receptors in network excitability and seizure susceptibility. Recent advances in molecular regulation of extrasynaptic GABA-A receptor-mediated tonic inhibition are providing unique new therapeutic approaches for epilepsy, status epilepticus, and certain brain disorders. The discovery of an extrasynaptic molecular mechanism represents a milestone for developing novel therapies such as NS replacement therapy for catamenial epilepsy.
Copyright © 2018 by The American Society for Pharmacology and Experimental Therapeutics.
Figures
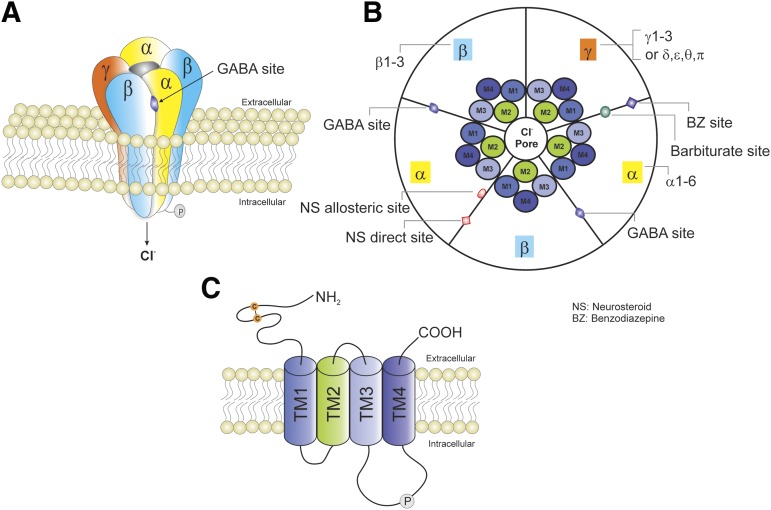
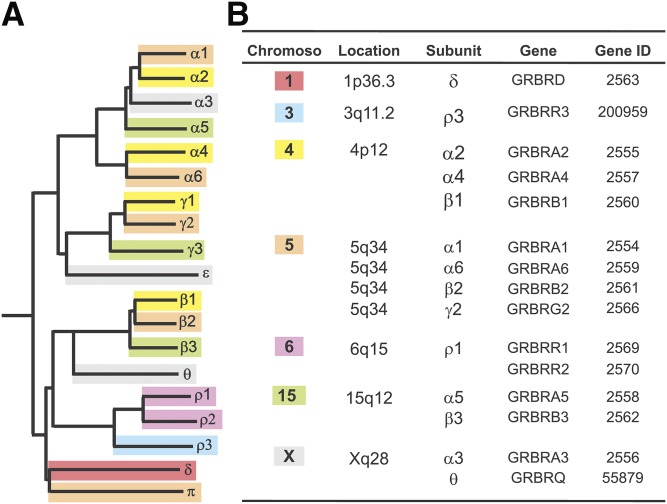
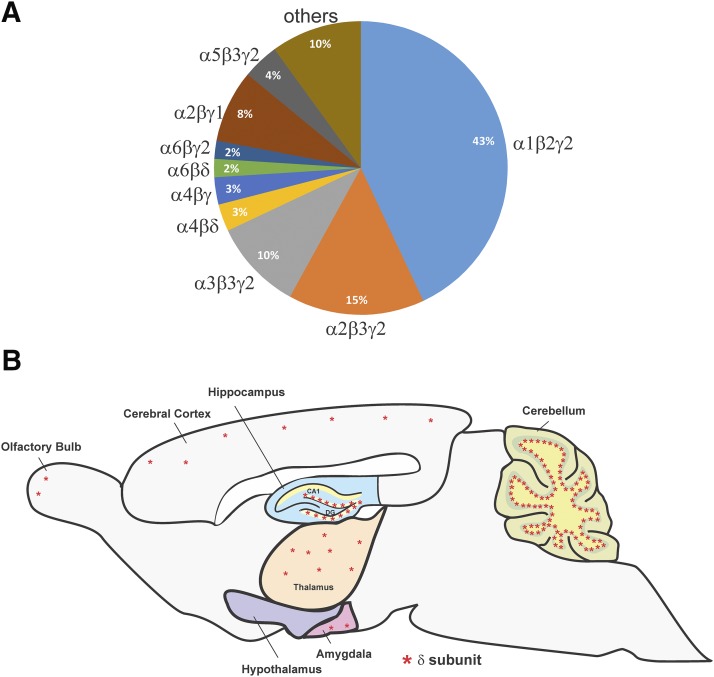
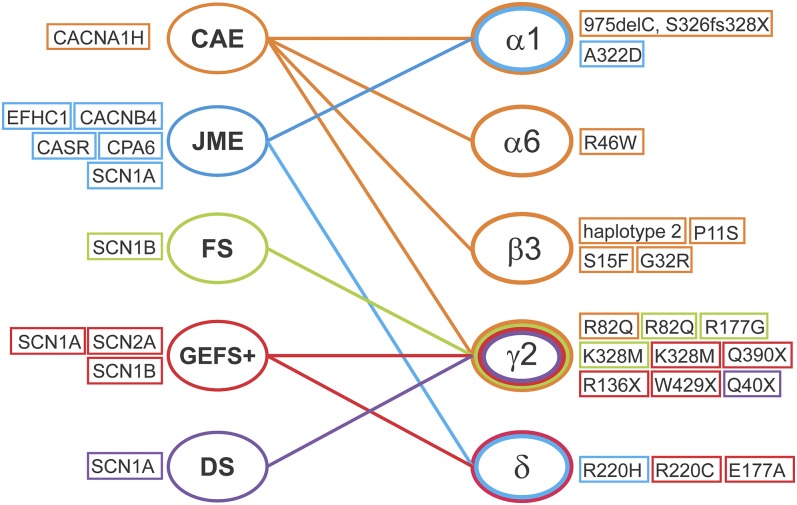
References
-
- Audenaert D, Schwartz E, Claeys KG, Claes L, Deprez L, Suls A, Van Dyck T, Lagae L, Van Broeckhoven C, Macdonald RL, et al. (2006) A novel GABRG2 mutation associated with febrile seizures. Neurology 67:687–690. - PubMed
-
- Baulac S, Huberfeld G, Gourfinkel-An I, Mitropoulou G, Beranger A, Prud’homme JF, Baulac M, Brice A, Bruzzone R, LeGuern E. (2001) First genetic evidence of GABA(A) receptor dysfunction in epilepsy: a mutation in the gamma2-subunit gene. Nat Genet 28:46–48. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
