

Novel mechanisms and functions of complement
- PMID: 29144501
- PMCID: PMC5706779
- DOI: 10.1038/ni.3858
Novel mechanisms and functions of complement
Abstract
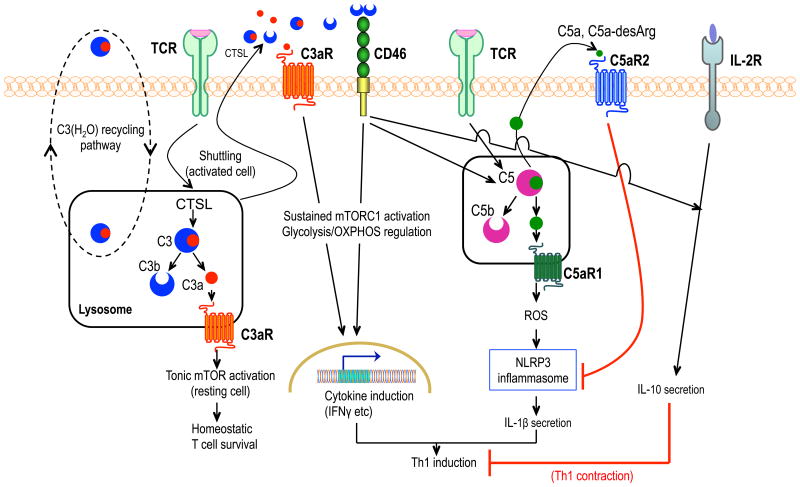
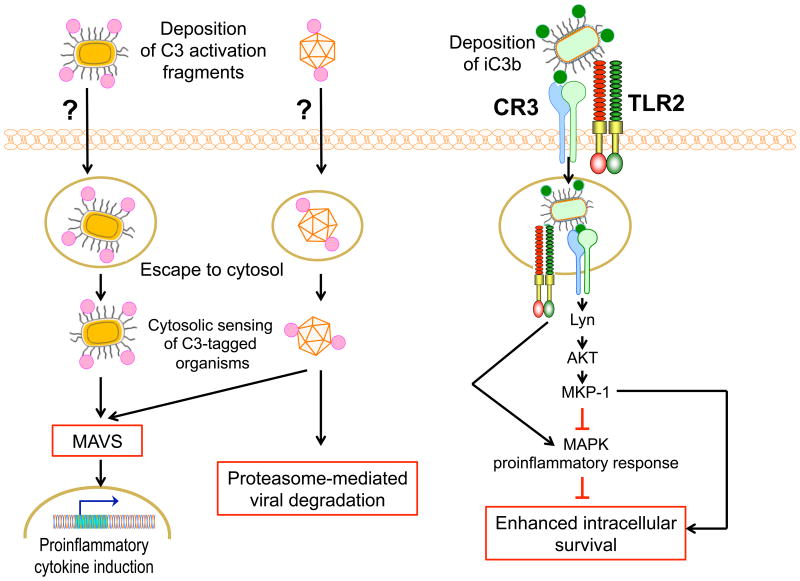
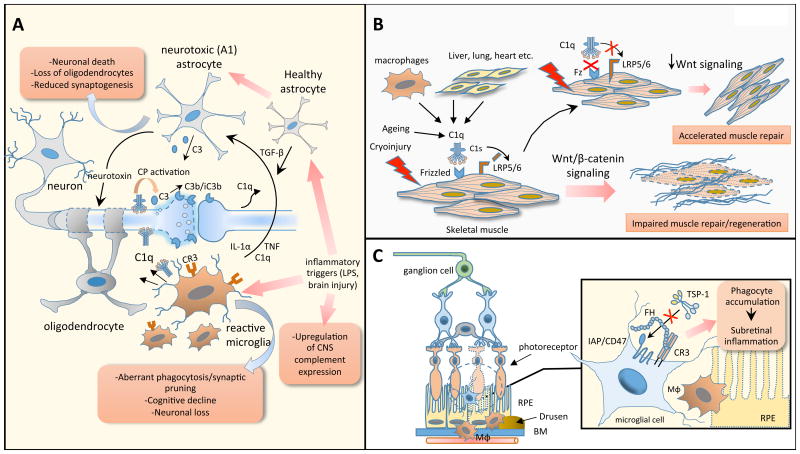
Progress at the beginning of the 21st century transformed the perception of complement from that of a blood-based antimicrobial system to that of a global regulator of immunity and tissue homeostasis. More recent years have witnessed remarkable advances in structure-function insights and understanding of the mechanisms and locations of complement activation, which have added new layers of complexity to the biology of complement. This complexity is readily reflected by the multifaceted and contextual involvement of complement-driven networks in a wide range of inflammatory and neurodegenerative disorders and cancer. This Review provides an updated view of new and previously unanticipated functions of complement and how these affect immunity and disease pathogenesis.
Figures


References
-
- Elmlund D, Le SN, Elmlund H. High-resolution cryo-EM: the nuts and bolts. Curr Opin Struct Biol. 2017;46:1–6. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
