

Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health
- PMID: 29145569
- PMCID: PMC5761609
- DOI: 10.1210/en.2017-00882
Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health
Abstract
The incidence of metabolic disorders like type 2 diabetes and obesity continues to increase. In addition to the well-known contributors to these disorders, such as food intake and sedentary lifestyle, recent research in the exposure science discipline provides evidence that exposure to endocrine-disrupting chemicals like bisphenol A and phthalates via multiple routes (e.g., food, drink, skin contact) also contribute to the increased risk of metabolic disorders. Endocrine-disrupting chemicals (EDCs) can disrupt any aspect of hormone action. It is becoming increasingly clear that EDCs not only affect endocrine function but also adversely affect immune system function. In this review, we focus on human, animal, and in vitro studies that demonstrate EDC exposure induces dysfunction of the immune system, which, in turn, has detrimental effects on metabolic health. These findings highlight how the immune system is emerging as a novel player by which EDCs may mediate their effects on metabolic health. We also discuss studies highlighting mechanisms by which EDCs affect the immune system. Finally, we consider that a better understanding of the immunomodulatory roles of EDCs will provide clues to enhance metabolic function and contribute toward the long-term goal of reducing the burden of environmentally induced diabetes and obesity.
Copyright © 2018 Endocrine Society.
Figures
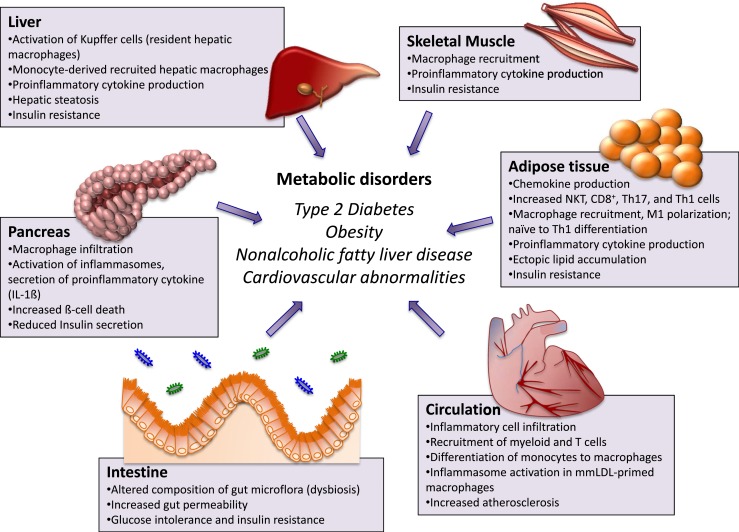
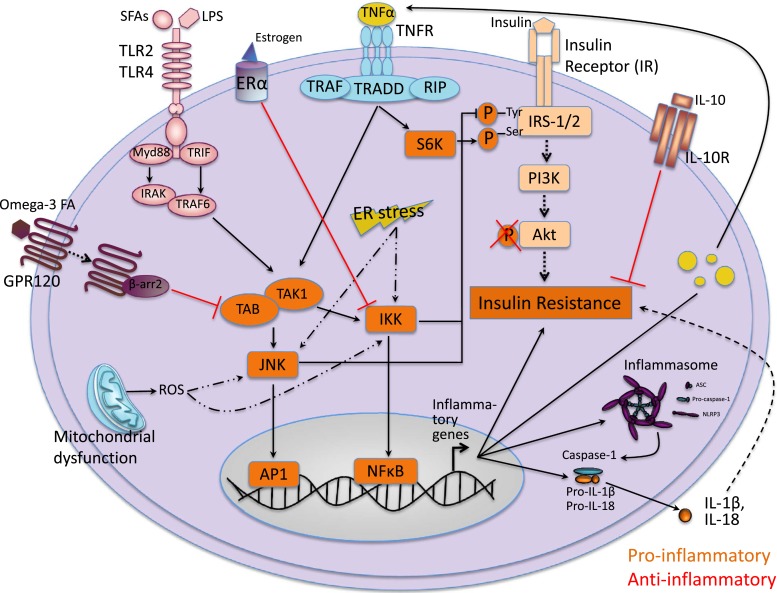
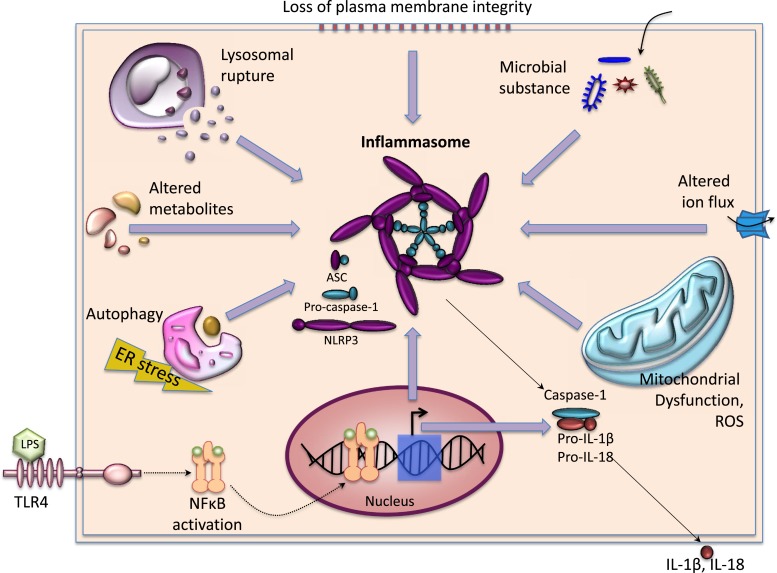
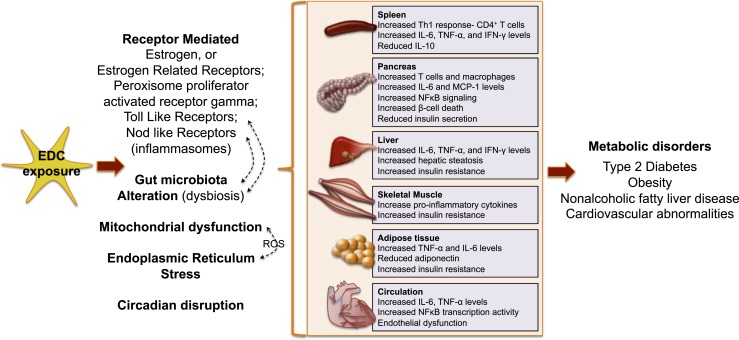
References
-
- Finkelstein EA, Khavjou OA, Thompson H, Trogdon JG, Pan L, Sherry B, Dietz W. Obesity and severe obesity forecasts through 2030. Am J Prev Med. 2012;42(6):563–570. - PubMed
-
- Newbold RR, Padilla-Banks E, Jefferson WN, Heindel JJ. Effects of endocrine disruptors on obesity. Int J Androl. 2008;31(2):201–208. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
