

Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics
- PMID: 29146941
- PMCID: PMC5691208
- DOI: 10.1038/s41598-017-15793-9
Developmental excitatory-to-inhibitory GABA-polarity switch is disrupted in 22q11.2 deletion syndrome: a potential target for clinical therapeutics
Abstract
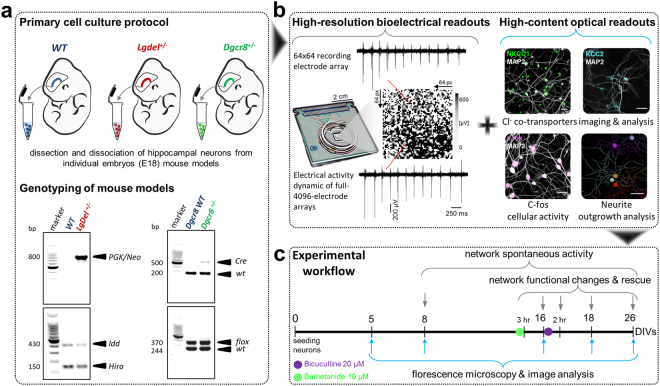
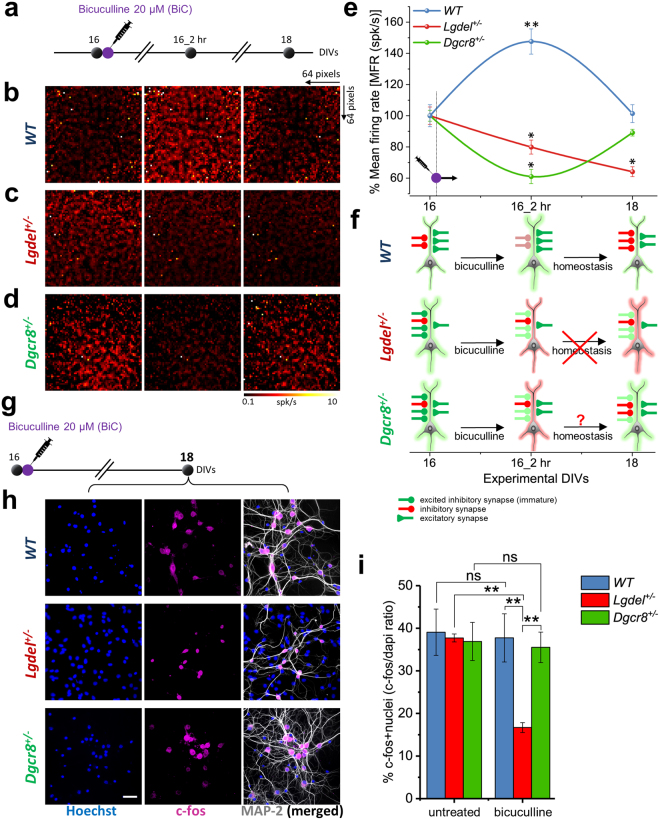
Individuals with 22q11.2 microdeletion syndrome (22q11.2 DS) show cognitive and behavioral dysfunctions, developmental delays in childhood and risk of developing schizophrenia and autism. Despite extensive previous studies in adult animal models, a possible embryonic root of this syndrome has not been determined. Here, in neurons from a 22q11.2 DS mouse model (Lgdel +/-), we found embryonic-premature alterations in the neuronal chloride cotransporters indicated by dysregulated NKCC1 and KCC2 protein expression levels. We demonstrate with large-scale spiking activity recordings a concurrent deregulation of the spontaneous network activity and homeostatic network plasticity. Additionally, Lgdel +/- networks at early development show abnormal neuritogenesis and void of synchronized spontaneous activity. Furthermore, parallel experiments on Dgcr8 +/- mouse cultures reveal a significant, yet not exclusive contribution of the dgcr8 gene to our phenotypes of Lgdel +/- networks. Finally, we show that application of bumetanide, an inhibitor of NKCC1, significantly decreases the hyper-excitable action of GABAA receptor signaling and restores network homeostatic plasticity in Lgdel +/- networks. Overall, by exploiting an on-a-chip 22q11.2 DS model, our results suggest a delayed GABA-switch in Lgdel +/- neurons, which may contribute to a delayed embryonic development. Prospectively, acting on the GABA-polarity switch offers a potential target for 22q11.2 DS therapeutic intervention.
Conflict of interest statement
The authors declare that they have no competing interests.
Figures





References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
