

Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas
- PMID: 29148847
- PMCID: PMC5743003
- DOI: 10.1080/14728222.2018.1406924
Emerging therapeutic targets in myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas
Abstract
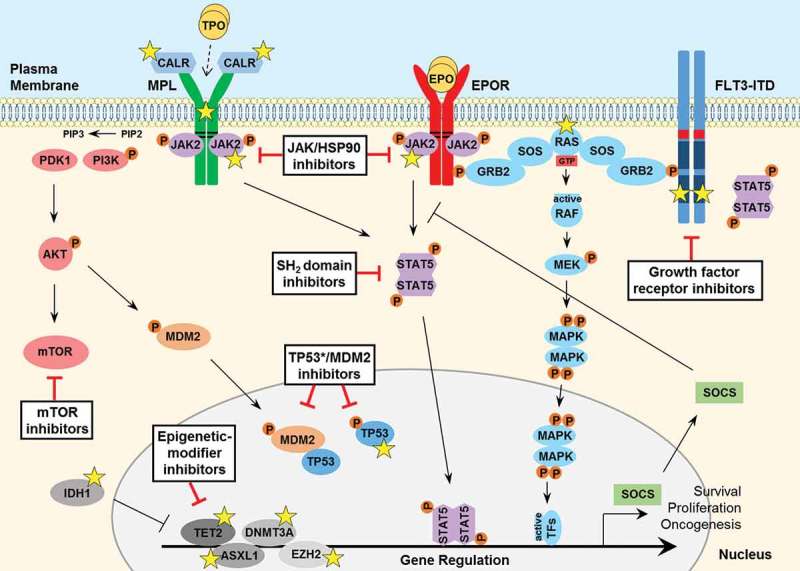
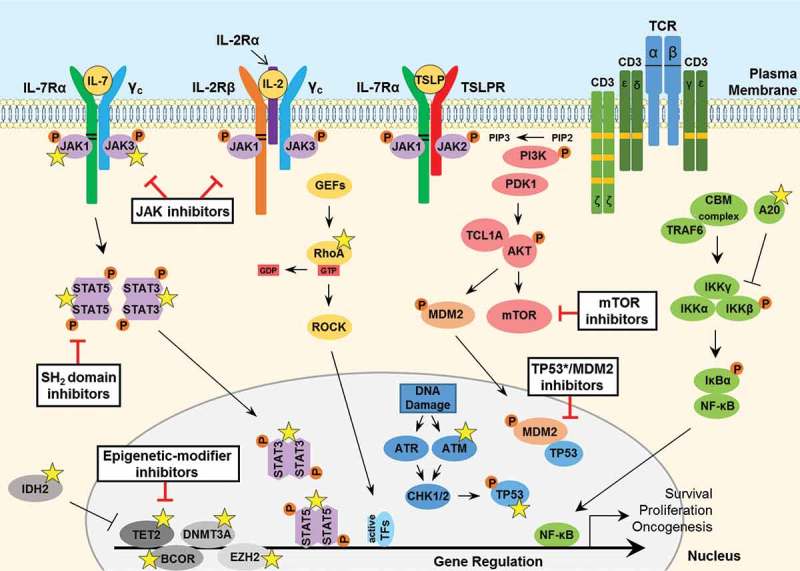
Hematopoietic neoplasms are often driven by gain-of-function mutations of the JAK-STAT pathway together with mutations in chromatin remodeling and DNA damage control pathways. The interconnection between the JAK-STAT pathway, epigenetic regulation or DNA damage control is still poorly understood in cancer cell biology. Areas covered: Here, we focus on a broader description of mutational insights into myeloproliferative neoplasms and peripheral T-cell leukemia and lymphomas, since sequencing efforts have identified similar combinations of driver mutations in these diseases covering different lineages. We summarize how these pathways might be interconnected in normal or cancer cells, which have lost differentiation capacity and drive oncogene transcription. Expert opinion: Due to similarities in driver mutations including epigenetic enzymes, JAK-STAT pathway activation and mutated checkpoint control through TP53, we hypothesize that similar therapeutic approaches could be of benefit in these diseases. We give an overview of how driver mutations in these malignancies contribute to hematopoietic cancer initiation or progression, and how these pathways can be targeted with currently available tools.
Keywords: Epigenetic target; MPN; PTCL; hematopoietic cancer; mutational landscape; therapeutic target.
Conflict of interest statement
The authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties. Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Figures
Similar articles
-
[Molecularly pathogenesis and molecular targeted therapy for myeloproliferative neoplasms].Rinsho Ketsueki. 2015 Feb;56(2):150-8. doi: 10.11406/rinketsu.56.150. Rinsho Ketsueki. 2015. PMID: 25765794 Review. Japanese.
-
JAK-STAT pathway activation in malignant and nonmalignant cells contributes to MPN pathogenesis and therapeutic response.Cancer Discov. 2015 Mar;5(3):316-31. doi: 10.1158/2159-8290.CD-14-0736. Epub 2015 Jan 8. Cancer Discov. 2015. PMID: 25572172 Free PMC article.
-
Rapid Molecular Profiling of Myeloproliferative Neoplasms Using Targeted Exon Resequencing of 86 Genes Involved in JAK-STAT Signaling and Epigenetic Regulation.J Mol Diagn. 2016 Sep;18(5):707-718. doi: 10.1016/j.jmoldx.2016.05.006. Epub 2016 Jul 19. J Mol Diagn. 2016. PMID: 27449473
-
A rare case report of 8p11 myeloid and lymphoid neoplasm with FGFR1 abnormality in a young adult.Ann Hematol. 2013 Jan;92(2):285-6. doi: 10.1007/s00277-012-1562-7. Epub 2012 Sep 2. Ann Hematol. 2013. PMID: 22941307 No abstract available.
-
New directions in treating peripheral T-cell lymphomas (PTCL): leveraging epigenetic modifiers alone and in combination.Expert Rev Hematol. 2019 Mar;12(3):137-146. doi: 10.1080/17474086.2019.1583102. Epub 2019 Feb 27. Expert Rev Hematol. 2019. PMID: 30782038 Review.
Cited by
-
Clinicopathological and molecular genomic features of monomorphic epitheliotropic intestinal T-cell lymphoma in the Chinese population: a study of 20 cases.Diagn Pathol. 2021 Dec 12;16(1):114. doi: 10.1186/s13000-021-01173-5. Diagn Pathol. 2021. PMID: 34895266 Free PMC article.
-
STAT5 as a Key Protein of Erythropoietin Signalization.Int J Mol Sci. 2021 Jul 1;22(13):7109. doi: 10.3390/ijms22137109. Int J Mol Sci. 2021. PMID: 34281163 Free PMC article. Review.
-
Overcoming the blood-brain barrier for the therapy of malignant brain tumor: current status and prospects of drug delivery approaches.J Nanobiotechnology. 2022 Sep 15;20(1):412. doi: 10.1186/s12951-022-01610-7. J Nanobiotechnology. 2022. PMID: 36109754 Free PMC article. Review.
-
Dual specific STAT3/5 degraders effectively block acute myeloid leukemia and natural killer/T cell lymphoma.Hemasphere. 2024 Nov 28;8(12):e70001. doi: 10.1002/hem3.70001. eCollection 2024 Dec. Hemasphere. 2024. PMID: 39619245 Free PMC article.
-
STAT5A and STAT5B-Twins with Different Personalities in Hematopoiesis and Leukemia.Cancers (Basel). 2019 Nov 4;11(11):1726. doi: 10.3390/cancers11111726. Cancers (Basel). 2019. PMID: 31690038 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous