

DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways
- PMID: 29149759
- PMCID: PMC5691221
- DOI: 10.1016/j.redox.2017.11.004
DUSP1 alleviates cardiac ischemia/reperfusion injury by suppressing the Mff-required mitochondrial fission and Bnip3-related mitophagy via the JNK pathways
Abstract
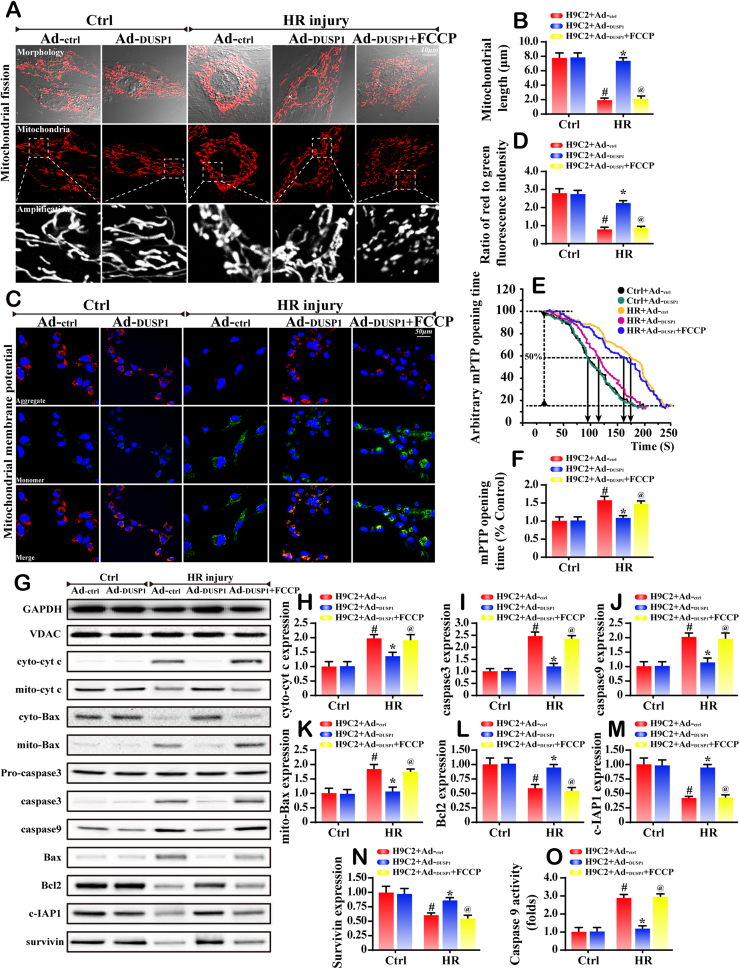
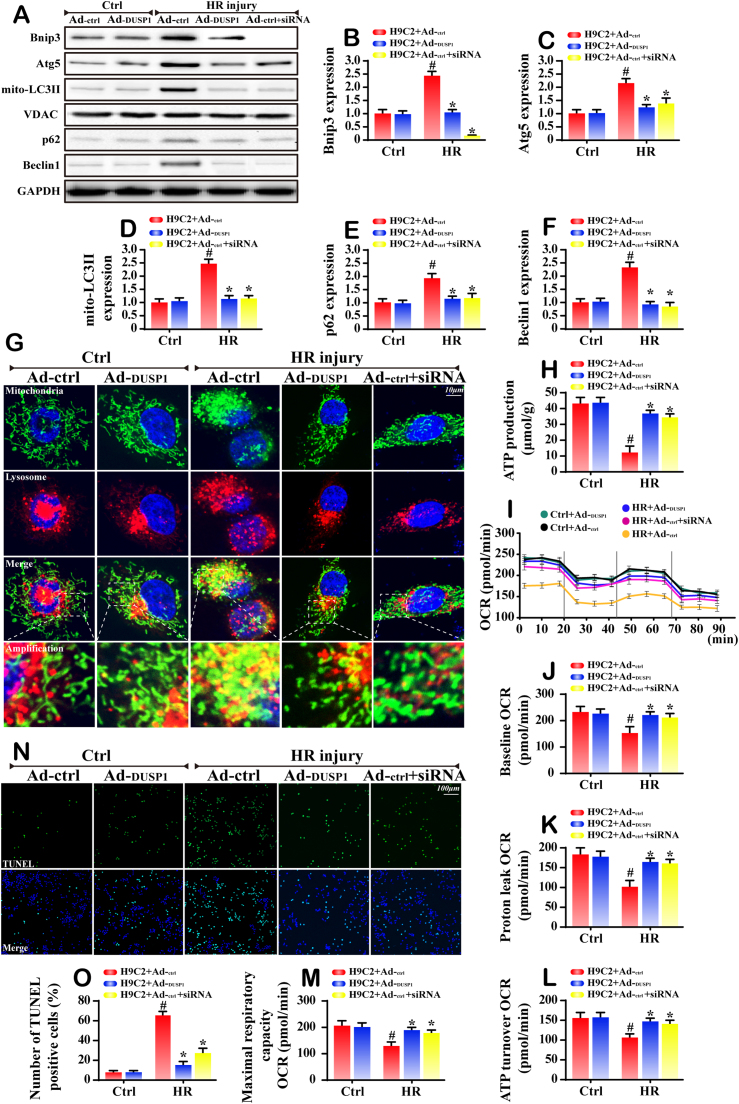
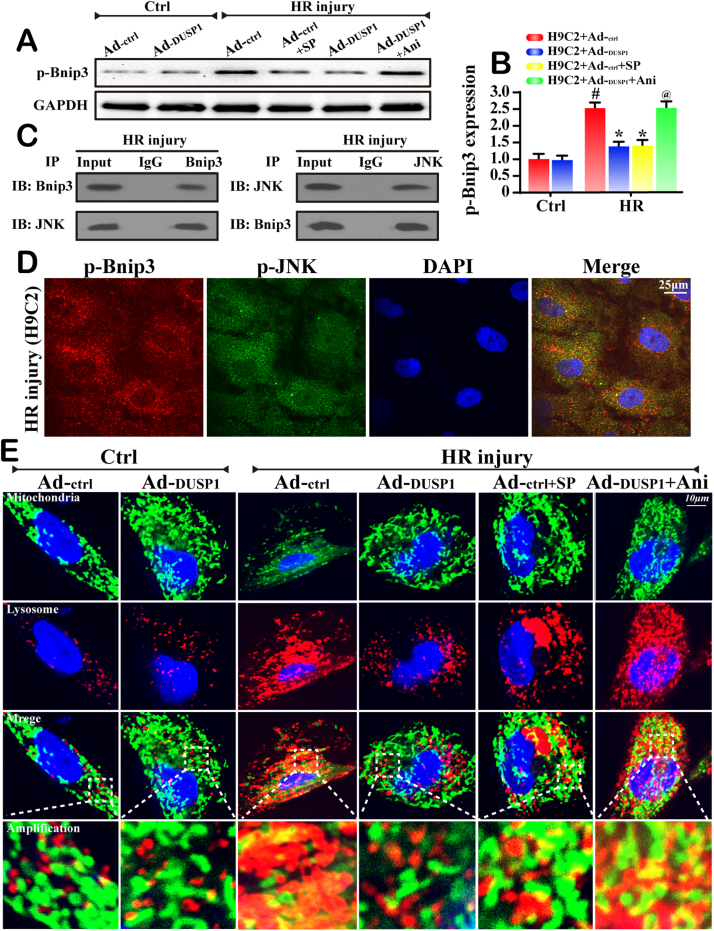
Mitochondrial fission and selective mitochondrial autophagy (mitophagy) form an essential axis of mitochondrial quality control that plays a critical role in the development of cardiac ischemia-reperfusion (IR) injury. However, the precise upstream molecular mechanism of fission/mitophagy remains unclear. Dual-specificity protein phosphatase1 (DUSP1) regulates cardiac metabolism, but its physiological contribution in the reperfused heart, particularly its influence on mitochondrial homeostasis, is unknown. Here, we demonstrated that cardiac DUSP1 was downregulated following acute cardiac IR injury. In vivo, compared to wild-type mice, DUSP1 transgenic mice (DUSP1TG mice) demonstrated a smaller infarcted area and the improved myocardial function. In vitro, the IR-induced DUSP1 deficiency promoted the activation of JNK which upregulated the expression of the mitochondrial fission factor (Mff). A higher expression level of Mff was associated with elevated mitochondrial fission and mitochondrial apoptosis. Additionally, the loss of DUSP1 also amplified the Bnip3 phosphorylated activation via JNK, leading to the activation of mitophagy. Increased mitophagy overtly consumed mitochondrial mass resulting into the mitochondrial metabolism disorder. However, the reintroduction of DUSP1 blunted Mff/Bnip3 activation and therefore alleviated the fatal mitochondrial fission/mitophagy by inactivating the JNK pathway, providing a survival advantage to myocardial tissue following IR stress. The results of our study suggest that DUSP1 and its downstream JNK pathway are therapeutic targets for conferring protection against IR injury by repressing Mff-mediated mitochondrial fission and Bnip3-required mitophagy.
Keywords: Bnip3; Cardiac IR injury; DUSP1; JNK; Mff; Mitochondrial fission; Mitophagy.
Copyright © 2017 The Authors. Published by Elsevier B.V. All rights reserved.
Figures





References
-
- Chen W.R., Chen Y.D., Tian F., Yang N., Cheng L.Q., Hu S.Y., Wang J., Yang J.J., Wang S.F., Gu X.F. Effects of liraglutide on reperfusion injury in patients with ST-segment-elevation myocardial infarction. Circ. Cardiovasc Imaging. 2016;9(12) - PubMed
-
- Chen W.R., Hu S.Y., Chen Y.D., Zhang Y., Qian G., Wang J., Yang J.J., Wang Z.F., Tian F., Ning Q.X. Effects of liraglutide on left ventricular function in patients with ST-segment elevation myocardial infarction undergoing primary percutaneous coronary intervention. Am. Heart J. 2015;170(5):845–854. - PubMed
-
- Zhou H., Hu S., Jin Q., Shi C., Zhang Y., Zhu P., Ma Q., Tian F., Chen Y. Mff-dependent mitochondrial fission contributes to the pathogenesis of cardiac microvasculature ischemia/reperfusion injury via induction of mROS-mediated cardiolipin oxidation and HK2/VDAC1 disassociation-involved mPTP opening. J. Am. Heart Assoc. 2017;6(3) - PMC - PubMed
-
- Bravo-San Pedro J.M., Kroemer G., Galluzzi L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 2017;120(11):1812–1824. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
